
Abstract
Adipose tissue (fat) is a heterogeneous organ, both in function and histology, distributed throughout the body. White adipose tissue, responsible for energy storage and more recently found to have endocrine and inflammation-modulatory activities, was historically thought to be the only type of fat present in adult humans. The recent demonstration of functional brown adipose tissue in adults, which is highly metabolic, shifted this paradigm. Additionally, recent studies demonstrate the ability of white adipose tissue to be induced toward the brown adipose phenotype – “beige” or “brite” adipose tissue – in a process referred to as “browning.” While these adipose tissue depots are under investigation in the context of obesity, new evidence suggests a maladaptive role in other metabolic disturbances including cancer-associated cachexia, which is the topic of this review. This syndrome is multifactorial in nature and is an independent factor associated with poor prognosis. Here, we review the contributions of all three adipose depots – white, brown, and beige – to the development and progression of cancer-associated cachexia. Specifically, we focus on the local and systemic processes involving these adipose tissues that lead to increased energy expenditure and sustained negative energy balance. We highlight key findings from both animal and human studies and discuss areas within the field that need further exploration.
Impact statement
Cancer-associated cachexia (CAC) is a complex, multifactorial syndrome that negatively impacts patient quality of live and prognosis. This work reviews a component of CAC that lacks prior discussion: adipose tissue contributions. Uniquely, it discusses all three types of adipose tissue, white, beige, and brown, their interactions, and their contributions to the development and progression of CAC. Summarizing key bench and clinical studies, it provides information that will be useful to both basic and clinical researchers in designing experiments, studies, and clinical trials.
Keywords
Introduction: The clinical problem – CAC
History, definition, and staging
Changes in metabolism and altered energy requirements have been documented in patients with cancer as early as the 1950s, 1 yet it was not until the 1970s that the multifaceted nature of CAC (also referred to as cancer cachexia) was appreciated.2,3 While early studies suggested that CAC was the result of tumor-driven anorexia alone, 4 it is now established that CAC is not solely the result of decreased food intake; other contributing factors such as malabsorption and altered metabolism play a role as well.2,3 In the early 2000s, increased emphasis was placed on consolidating clinical and basic research findings to develop guidelines for defining, staging, and treating CAC.
CAC, according to a recent international consensus developed by Fearon and Strasser, is defined as a “multifactorial syndrome defined by an ongoing loss of skeletal muscle mass (with or without loss of fat mass).” 5 Almost all cancers can be associated with CAC, although some more commonly than others. 6 Classically, CAC is described in three stages: precachexia, cachexia, and refractory cachexia. Several assessment criteria were proposed and described in detail by Fearon et al. 5 ; the most obvious is the degree of weight loss a patient experiences, but others include catabolic drive and loss of muscle mass and strength. The consensus guidelines were quickly applied to and validated in clinical practice, and additional assessment and management algorithms were proposed.7,8
While these guidelines represent significant improvements, there is still a lack of simple clinical indicators and markers to allow for earlier and straightforward classification of CAC. 8 Despite increased awareness of this condition, CAC is still very common and treatments are inadequate, 9 suggesting that a comprehensive understanding of the molecular and physiologic pathways leading to the development and progression of CAC is still lacking. The large inter-individual variability in the prevalence and severity of CAC in patients with the same tumor type10,11 further supports the need for improving the knowledge of the pathophysiologic mechanisms responsible for CAC. Given the high prevalence of obesity in the general population, early stages of CAC may be missed when the diagnosis is based purely on weight loss criteria (referred to as the “obesity paradox” 11 ). This factor demonstrates the need for improvement in the early diagnosis of this condition.
Prognosis, effects, and treatment options
The inverse association between weight loss and survival in patients with cancer is well known. 12 Recent estimates propose that CAC affects 60–80% of all patients with advanced cancer, 8 and is directly implicated in at least 20% of cancer-related deaths, 13 demonstrating that CAC is an independent risk factor for poor prognosis. The loss of body mass and altered body composition in CAC leaves patients vulnerable to increased toxicity from anti-tumor therapies such as chemotherapy.8,14,15 This may directly result in increased morbidity and mortality, and may indirectly affect morbidity and mortality as the increased toxicity requires clinical treatment delays and dose reductions in therapy. Not surprisingly, patients with CAC additionally experience loss of physical function and decreased quality of life.8,14
Clinical advancements in defining and staging CAC have not been matched by similar improvements in treatment options and patient outcomes. Some studies suggest that CAC may be reversible if caught in the precachexia or cachexia stages.7,15 The main therapeutic strategies utilized in these stages include exercise, intense nutritional support, and removing any direct tumor causes (such as obstruction causing malabsorption).7,8 Unfortunately, many cases are clinically diagnosed in the refractory cachexia stage, when CAC is often irreversible5,7 and nutritional support, current drug therapies, and other measures are ineffective in restoring a net-neutral or positive energy balance.16,17 In the past, many studies investigating therapeutic drugs to combat CAC, such as eicosapentaenoic acid diester 18 and bortezomib 19 have failed to demonstrate favorable outcomes. While experiments and studies investigating new potential therapeutic targets have been ongoing, when Fearon published his article in 2011, there were no active clinical drug trials for CAC 5 ; since then, several drugs in the pipeline have entered into the clinical trial phases. A review by Dingemans et al. 20 identified 12 phase II clinical trials with 11 compounds. Each of these drugs are expected to help combat CAC via one of the following mechanisms: increasing appetite, improving digestion, decreasing systemic inflammation, and improving the muscle synthesis-versus-degradation ratio. 20 Other drugs have recently entered phase III trials, but difficulties in meeting multiple clinical endpoints indicate the need for a more comprehensive approach. Anamorelin, a ghrelin-receptor agonist, demonstrated an ability to improve lean body mass in a phase II clinical trial with patients with non-small cell lung cancer and CAC. 21 However, this phase II trial, and a subsequent phase III trial, reported that while lean body mass improved, functional improvements in hand grip strength did not21,22 – highlighting the potential discrepancy between proxies of end-organ effect and patient-centered significant outcomes. Enobosarm, a selective androgen receptor modulator, showed a significant increase in total lean body mass in phase II studies, 23 but according to information thus far presented in abstract format, it has not yielded consistent endpoint results in phase III trials.24,25 As described below, there are many contributing factors to CAC, so the development of these drugs is certainly a step in the right direction. However, few compounds make it past phase II and III studies, and those that have (and will) may not fully address the spectrum of pathophysiologic components of CAC to be effective in mitigating or reversing all of the clinical components of CAC.
Contributing factors
The factors contributing to the development and maintenance of CAC are grounded in the axiom of energy balance: weight loss or gain only occurs when there is a sustained imbalance between energy intake and energy expenditure.3,26 In the case of CAC, there is a persistent net-negative energy balance, with components linked to both decreased energy intake and increased energy expenditure. Decreased energy intake may occur via several mechanisms, including anorexia caused by chemosensory distortions, malabsorption, and early satiety.5,7,10,14 Increased energy expenditure may result from inflammation, increased tumor metabolism, and altered/increased metabolism.7,10,11,16 To this end, the heterogeneity of adipose tissue is particularly relevant in the pathophysiology of CAC. Adipose tissue metabolism, aside from its more obvious role in obesity and diabetes, has been demonstrated to play a role in other states of metabolic dysfunction such as CAC. As such, this minireview will identify and discuss the specific contributions of adipose tissue to the development and progression of CAC.
Adipose tissue: A heterogeneous and endocrine organ
Adipose tissue, previously viewed as an inert fat depot in isolated areas within the body, is now regarded as a large, interactive, multi-compartment organ with clear organization and anatomy.27–29 Additionally, the secretomes of these depots can act locally (paracrine organ function) and systemically (endocrine organ function).30,31
White, brown, and beige adipose tissue
The largest component of adipose tissue is white adipose tissue (WAT), which primarily consists of large, spherical adipocytes with a unilocular lipid droplet consuming most of the cell volume. 29 WAT is found in both subcutaneous and visceral depots, and increased visceral WAT mass is associated with increased metabolic risk.32,33 WAT has important endocrine and paracrine roles throughout the body,29,34,35 and in general terms, functions to store energy, mainly in the form of triglycerides.36,37
In contrast to WAT, brown adipose tissue (BAT) primarily functions to expend energy. 36 Until recently, functionally significant BAT was thought to be present only in neonates, undergoing rapid involution with age,36,38–41 despite early reports that indicated the presence of BAT in adult humans.39,40,42 The development and use of positron-emission tomography (PET) allowed for visualization of BAT in adult humans, 43 and in combination with other functional analyses,44,45 sparked the recent surge in interest in BAT’s function in health and disease. Details of WAT and BAT development and gene signature are discussed elsewhere,36,46,47 but it is important to note that, at least in mice, anatomically defined BAT adipocytes are derived from a cell lineage different from WAT, which instead share a lineage with myocytes (muscle cells).48,49 Adult human BAT depots are located near the aorta and within the supraclavicular region of the neck. 29 These adipocytes contain multilocular lipid droplets, and their ability to expend uniquely great amounts of energy is largely due to the presence and activation of a proton leakage pathway 50 mediated by uncoupling protein 1 (UCP1) – the hallmark of BAT function.39,51 UCP1 uncouples oxidative phosphorylation from ATP synthesis in the inner mitochondrial membrane to dissipate energy in the form of heat.52,53
More recently, an additional type of adipocyte, defined as “brite” (brown-in-white) 54 or “beige” 55 was identified. These adipocytes are located within and share cell lineage with white adipocytes 36 but express UCP1 and function like brown adipocytes. 54 While some authors suggest that beige cells develop de novo, 56 the model of trans-differentiation – development of beige adipocytes from pre-existing white adipocytes 57 – predominates. 29 Interestingly, in addition to sustained exposure to cold, multiple different pathways promote the white-to-beige transition leading to a similar cellular phenotype, reviewed by Giralt and Villarroya. 36 Importantly, beige adipose tissue can expand (“plasticity”) in response to several mediators and is a target of both endocrine and paracrine stimuli. 58
This relatively “new” beige adipose tissue has generated a great deal of excitement, as its function is similar to classical BAT in expending energy and may have additional protective roles against obesity. 36 On the other hand, its capability of developing, expanding, and activating under local stimuli, including extracellular matrix components, 59 make this tissue a potential driver of CAC in response to tumor microenvironment. Together, all three adipose tissue types are clearly important in the energy balance that is disrupted in CAC.
Adipose tissue contributions to CAC
Lipolysis
The breakdown of adipose tissue – lipolysis – is perhaps the most evident component of adipose tissue’s contribution to CAC. 60 Fat loss observed in patients with CAC is thought to occur via breakdown of adipose tissue (mainly WAT) in response to a negative energy balance due to cancer-associated anorexia and other pathologic factors recently reviewed by Ebadi and Mazurak.61,62 The importance of lipolysis in CAC was demonstrated in a study that showed that the inhibition of lipid mobilization can improve the CAC state. 63 A murine model of animals implanted with murine adenocarcinoma 16 (MAC16) tumors reported changes in adipose tissue that included shrunken adipocytes and decreased expression of adipose tissue transcription factors. 64 From a clinical perspective, a critical component of the body mass loss observed in CAC is the depletion of muscle mass, which usually precedes the observation of significant changes in adipose tissue mass and is associated with decrease in muscle function and mobility. Fearon et al.’s 5 consensus findings highlight that skeletal muscle loss is a necessity for a clinical diagnosis of CAC, but that adipose tissue loss may or not be present. However, lipolysis and lipid wasting may occur to an extent before muscle loss. 63 Additionally, studies in a mouse model of colon cancer demonstrated an increase in protein kinase-A-mediated lipolysis in early stage cachexia, 65 and this “early” lipolysis was implicated in (1) the inception of a negative energy balance that worsens over the course of CAC progression, and (2) a direct loss of skeletal muscle. 66 Lipolysis results in increased free fatty acids in circulation, which then get taken up by skeletal muscle; the excess of intramuscular free fatty acids results in several biochemical changes, such as the expression of ubiquitin lipases Atrogin-1 and MuRF 67 that lead to skeletal muscle atrophy. 68 Indeed, Stephens et al. 69 showed a positive association between the extent of body weight loss in cancer patients and the amount of lipid droplet accumulation within skeletal muscle cells. In a study of later-stage CAC, protein kinase-A-mediated lipolysis was not observed as in early stages, but instead more lipases were observed and contributed to skeletal muscle dysfunction and atrophy. 70 These findings suggest that there are likely different stages of lipid metabolic responses in CAC. Physiologically, one would expect that lipolysis and resulting loss of WAT mass would stimulate other pathways in the body to drive anabolism and energy intake, such as leptin. Leptin is produced by WAT and its levels are positively correlated to a patient’s state of adiposity, regardless of age or BMI. 61 Low levels of leptin are expected in patients exhibiting fat loss with CAC, which should result in increased activation of orexigenic pathways; however, studies demonstrate that this feedback may be disrupted in cancer, resulting in an undesirable decrease in signaling pathways such as neuropeptide Y. 71 It is important to recognize that inflammation is known to be a key player in lipolysis and the general increased catabolic drive observed in CAC (reviewed by Penet and Bhujwalla 16 ). Since adipose tissue depots have interspersed lymphocytes 29 and macrophages, 72 there may be a functional relationship between lipolysis and increased inflammation. However, CAC can present even in the absence of frank systemic inflammation, 5 and therapies targeting inflammatory cytokines such as tumor necrosis factor-alpha and eicosapentaenoic acid diester have not been successful in ameliorating CAC.18,73 This finding is consistent with the multifactorial nature of CAC.
Brown and beige adipose tissue expansion and activation
Several studies highlight increased resting energy expenditure in animal models as well in humans affected by CAC, and point to adipose tissue as a culprit.74–76 While lipolysis literally represents a loss of adipose tissue (especially WAT) mass, BAT and beige adipose tissue depots appear to contribute to CAC via increased energy expenditure.
BAT was first proposed as a contributor to CAC in 1989, 77 but this hypothesis was not investigated further until the rediscovery of functional BAT in adult humans. 44 In a mechanistic study, Tsoli et al. 74 found that activation of BAT (via increased Ucp1 expression) contributed to the development of CAC in mice with cachectic colon cancer cell line injections. Interestingly, CAC and BAT activation were not present in mice with non-cachectic colon cancer cell line injections. 74 In another study, BAT activity was measured via 18F-fluorodeoxylucose positron-emission tomography/computed tomography (FDG PET/CT) in human patients with cancer and was found to correlate positively with cancer stage. 78 However, classical BAT is relatively limited in mass, localized to relatively small depots within the body, therefore severely limiting its capacity to offer substantial contributions to energy expenditure and CAC in humans.79,80 More studies are needed to provide a quantitative assessment of classical BAT’s contributions to CAC. 81
WAT, however, has much larger depots, so the expansion of beige adipose tissue within WAT depots may confer greater increases in energy expenditure and may contribute significantly to CAC. Not surprisingly, WAT browning was identified in patients with pheochromocytomas, 82 neuroendocrine tumors that secrete norepinephrine, a known trigger of white-to-beige trans-differentiation. However, WAT browning has also been observed in other tumor models beyond catecholamine-secreting tumors. Mechanistic studies in mice include genetic models of Kras-lung, Kras-pancreatic, and K5-SOS carcinomas, 83 chemically induced liver carcinoma, 83 graft/injection models of B16 melanoma 83 and Lewis lung carcinoma,83,84 C26 colon carcinoma with and without IL6 expression, 83 and human pancreatic carcinoma xenografts. 83 In these models, investigators reported the presence of WAT browning and its contribution to the observed CAC. This was demonstrated by increased expression of Ucp1 mRNA and UCP1 protein,83,84 and also through increased expression of other genes crucial for beige adipose tissue development, such as Prdm16 and Pgc1α. 84 The authors also performed functional analyses of the proposed beige adipose tissue to demonstrate increased energy expenditure using oxygen consumption rate assays as well as body weight, tissue weight, physical activity, oxygen uptake (VO2), carbon dioxide production (VCO2), respiratory exchange ratio (RER), and heat generation measurements.83,84 Additionally, two drivers of white-to-beige trans-differentiation were identified: interleukin-6 (IL-6) 83 and parathyroid hormone-related peptide (PTH-rp). 84 These are the first studies to identify causative agents secreted by tumors that contribute to WAT browning and result in increased energy expenditure, which contributes to CAC. Importantly, WAT browning was observed early in the progression of CAC, before skeletal muscle atrophy occurred (consistent with the observations in lipolysis studies63,65), suggesting that adipose tissue dysfunction may occur prior to clinically evident adipose tissue loss, contributing to the development of CAC. Furthermore, Petruzzelli et al. 83 examined, via immunohistochemistry, adipose tissue samples acquired from human patients with CAC and a multitude of cancers: Kaposi’s sarcoma, melanoma, cholangiocarcinoma, colon adenocarcinoma, pancreatic neuroendocrine cancer, pleomorphic lung carcinoma, and lung adenocarcinoma. 83 Results showed that many of the WAT samples had increased UCP1 expression and adipocyte atrophy. While there is still much to understand, these results clearly support the role of WAT browning in the development and progression of CAC.
Where are we headed?
Addressing the gaps in knowledge
The current understanding of CAC, while certainly growing, is still not sufficient, and a complete picture of the role of adipose tissue in CAC is missing. As previously mentioned, the potential for dynamic changes in lipolysis and brown and beige adipose tissue function throughout the course of CAC must be investigated further. The interplay between lipolysis and beige and BAT is also an important, but not well understood, component. Quantification of the capacity of BAT activation to increase energy expenditure in patients with CAC will be helpful to understand the actual contribution of this tissue to the observed negative energy balance.
The least understood area with the greatest need for further research in this context is WAT browning. While morphologic studies identifying the presence of browning are clearly important, additional effort should be devoted to develop better models to investigate specific mechanisms involved in this process. Even though some gastro-enteric malignancies are more often associated with CAC, additional cancer models which provide direct contiguity to WAT and are removed from other factors which may confound mechanistic studies (for example, malabsorption due to mechanical obstruction) should be considered. Additionally, as more experiments are designed, mouse housing conditions and thermoneutrality must be taken into consideration, since cold exposure is a potent stimulator of WAT browning, 85 and tumor growth and inflammation are exacerbated when mice are housed at sub-thermoneutral temperatures. 86 These technical factors cannot be overlooked when designing or interpreting experiments in order to decrease confounding contributions to increased energy expenditure. Furthermore, the translation to human research is important but underrepresented. The experiments with human samples from the study discussed in the prior section 83 looked at samples stained for UCP1, but no mechanistic studies or functional analyses were performed. By improving experimental design to address these technical shortcomings, translational researchers will be better suited to succeed in developing novel therapeutic modalities aimed to mitigate the effects of CAC.
As more mechanistic information about WAT browning under non-cancer conditions is elucidated, more potential targets in CAC may emerge. For example, fibroblast growth factor-21 (FGF21) has been shown to not only be a target for BAT activation and thermogenesis, but may also be secreted by beige and brown adipocytes (for review, see Ni et al. 87 ). Molecules such as FGF21 may be relevant in the context of CAC and should be investigated. Although inflammation plays a role in global increased catabolism, and to an extent the WAT browning observed in some models, CAC can also occur without overt inflammation. 5 As such, it is possible that like PTH-rp, other drivers of browning not directly part of the inflammatory response play a causative role. Again, the development of better models and mechanistic studies will allow for elucidation of this information.
The other side of the coin: The role of adipose tissue in promoting cancer progression
While this review is focused on the effects of adipose tissue on CAC due to the presence of cancer, it is important to recognize that the adipose tissue–cancer interactions also go in the opposite direction. Cancer may directly and indirectly affect adipose tissue, and adipose tissue may directly and indirectly affect cancer growth and survival, as well. 88 Recent studies demonstrated that adipose-derived fibroblasts and cancer-associated adipocytes can contribute to cancer progression,30,89,90 and another study demonstrated that increased beige and BAT contributes to tumor progression. 91 Since higher rates of lipolysis and browning are strongly correlated with later-stage CAC, the hypothesized cross-talk between tumor and adipose tissue may generate a positive feedback loop facilitating cancer progression and the clinical impact of CAC.
Potential therapeutic strategies to restore adipose tissue homeostasis in CAC
As mentioned previously, aberrations in adipose tissue metabolism and homeostasis in malignancy result in a decrease in both adipose tissue and muscle mass. An ideal therapeutic approach would improve both adipose tissue and lean muscle mass; however, this has not been successful due to the differences in cell lineage of muscle and WAT. Their common precursor is the mesenchymal stem cell, but adipogenesis and myogenesis have very different differentiation signals and pathways, such as differences in Wnt signaling. 92 Differences are also observed mechanically, as softer substrates promote adipogenesis while stiffer substrates promote myogenesis. 93 These differences, and subsequent lack of overlapping triggers, make the development of a single therapeutic aimed at increasing both WAT and muscle mass incredibly difficult.
This relationship between adipose tissue changes and muscle loss is observed in other non-malignancy states as well: in postmenopausal women with type 2 diabetes mellitus, for example, increased intramuscular adipose was associated with hand grip strength. 94 In the elderly population, increased thigh muscle lipid content, represented by lower skeletal muscle attenuation coefficients on computed tomography images, was demonstrated to be negatively associated with muscle attenuation and strength. 95 Even in patients without metabolic disease, increased thigh intramuscular adipose tissue is associated with predictors of increased metabolic syndrome risk. 96 Current clinical trials investigating potential therapeutic agents for patients with CAC are promising, but the drugs being tested do not directly address the contributions of white, beige, and BAT to CAC (such as reducing the release of free fatty acids from WAT, or inhibiting the white-to-beige transition). AR-42, a histone deacetylase, has recently demonstrated preclinical success in reducing levels of MuRF1 and Atrogin-1 lipases and decreasing muscle degradation. 97 So, while still mostly speculative, it is possible that restoring adipose tissue homeostasis by targeting these areas of dysfunction will contribute to further improvements in patient-centered outcomes. For example, inhibition of WAT lipolysis and its downstream effects on skeletal muscle atrophy could lead to improvement of hand grip strength (used as a proxy for overall muscle strength and function) and assessments of physical activity (usually via patient-reported rating of physical functioning). Patients’ self-reported quality of life and psychosocial status will likely improve if adipose tissue mass loss (and skeletal muscle, as well) was dampened – in part because patients may feel that they are in better control of their weight and can keep their strength, and also in part because conservation of adipose tissue mass would likely result in a decrease in anti-tumor therapy toxicities. While studies investigating a ketogenic diet demonstrate improved quality of life in patients with advanced cancer, 98 to our current knowledge, no studies have investigated the capability to restore adipose tissue mass and the subsequent effect of restoring adipose tissue mass on CAC. A comprehensive understanding of the physiology and pathophysiology of adipose tissue, cancer, and CAC will hopefully lead to the discovery of key components and drivers within the development and progression of CAC that can be manipulated and targeted to benefit the patient. This is not a simple task, but as research progresses, the future applicability to patients suffering from CAC must always be considered a primary endpoint.
Applicability to obesity
Clearly, the implications of research in white, beige, and BAT are not limited solely to CAC. Adipose tissue is a key player in other states of metabolic dysfunction, and the number of patients who are overweight, obese, and severely obese continues to rise.99,100 The metabolic dysfunctions classically associated with obesity (such as type 2 diabetes mellitus) have a severe negative impact on patient duration and quality of life. Pharmacologic interventions targeting adipose tissue to treat diabetes, obesity, and other related metabolic disorders continue to be designed, developed, and tested.101–103 It is important to keep in mind that whole body homeostasis is very complex, and while obesity and CAC fall on opposite extremes of the spectrum, they share some key similarities that make research in one area applicable to the other. Muller et al. 104 point out that changes in body composition in both CAC and obesity are tied to the masses of individual organs and tissues, relationships between individual organs and tissue masses, and cross-talk between organs and tissues. 104 For example, concurrent obesity and sarcopenia, termed “sarcopenic obesity,” are highly prevalent. 105 Pathophysiologic mechanisms of the muscle and strength loss observed in patients with sarcopenic obesity may overlap with mechanisms of the same phenotype in patients with CAC, and adipose tissue homeostasis could be a common key factor. Therefore, it is likely that as the interactions between adipose tissue and cancer are elucidated in the context of increased energy expenditure, some of the findings and targets may lead to novel and effective therapeutic tools to treat or prevent the development of obesity and its metabolic consequences.
Conclusion
CAC is a severe and life-limiting complication of various forms of cancer. Adipose tissue’s role in CAC has evolved from a passive loss of energy stores due to reduced intake to an active process, with a multitude of drivers of its development and progression. Mechanisms, illustrated in Figure 1, include the metabolic disturbances due to unrestrained lipolysis, and increased energy expenditure due to beige and BAT expansion and activation. Furthermore, maladaptive expansion of beige adipose tissue may promote cancer progression. Additional research will allow for a better understanding of the molecular mechanisms leading to CAC and could in turn enable the development of therapeutic interventions to improve this condition. Conversely, the expanded knowledge of CAC pathophysiology may provide novel treatment options to promote energy dissipation, leading to amelioration of obesity and its metabolic complications.
A summary of adipose tissue contributions to cancer-associated cachexia (CAC). Cancer and its microenvironment (top) influence white, beige, and brown adipose tissue (colored respectively). WAT undergoes lipolysis, which results in a direct loss of adipose tissue mass and also contributes to skeletal muscle mass loss. As the process progresses, skeletal muscle loss may act as positive feedback for further adipose tissue lipolysis. WAT may also undergo browning to undergo trans-differentiation to beige adipose tissue, expressing uncoupling protein 1 (UCP1) and thereby expending greater amounts of energy. Similarly, existing classical brown adipose tissue may be activated, resulting in greater UCP1 expression with a resulting increase in energy expenditure. Collectively, these changes result in a net-negative energy balance, which contributes to the development and progression of CAC. (A color version of this figure is available in the online journal.)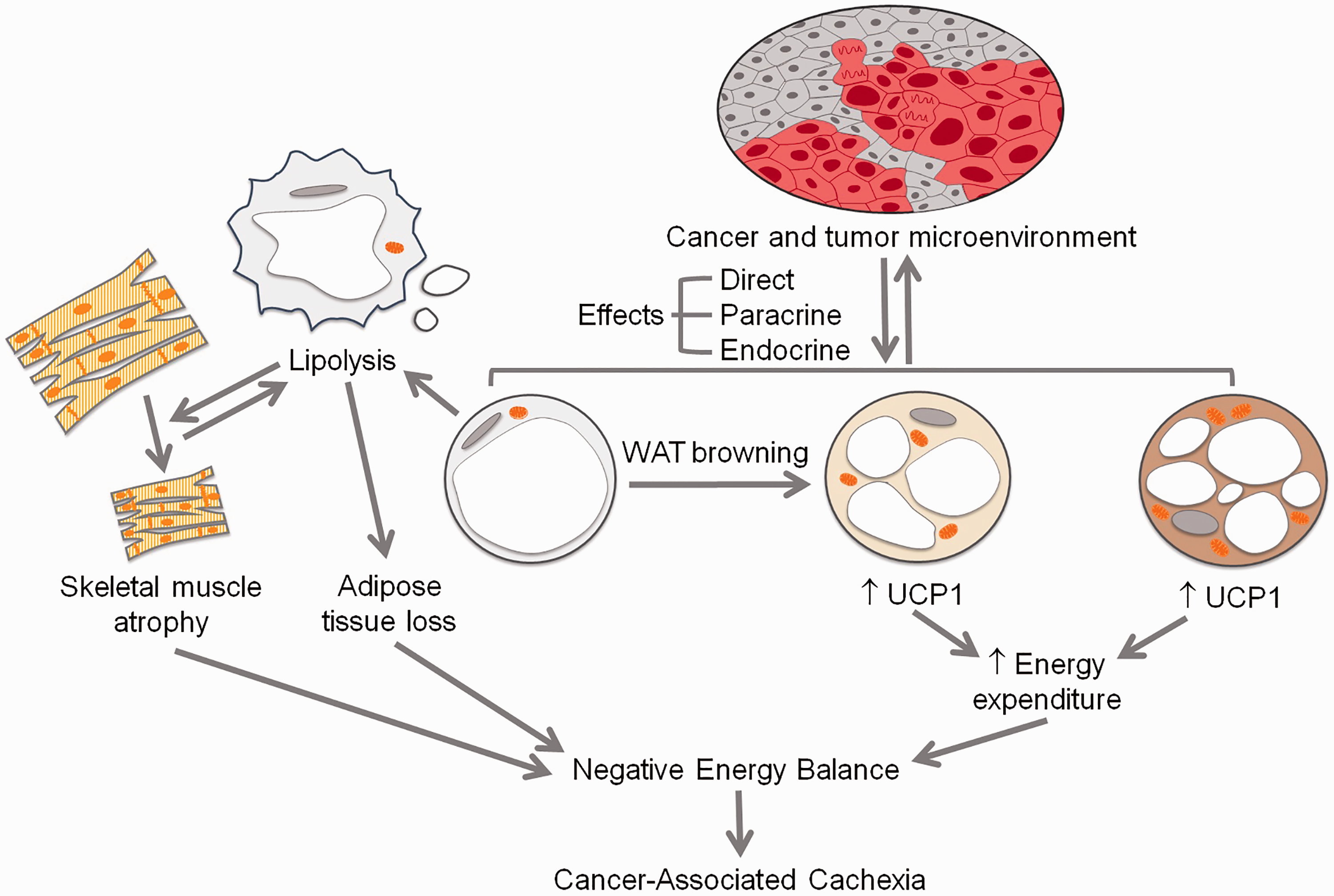
Footnotes
Authors’ contributions
JAV wrote the manuscript, with significant contributions and guidance from FSC.
Acknowledgments
The authors would like to thank Jared S. Farrar for his helpful edits to the final version of this manuscript. Additionally, the authors would like to thank the American Physician Scientist’s Association and the Society of Experimental Biology and Medicine for the opportunity to submit this minireview.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
There was no funding to support this minireview.