
Abstract
Ischemic preconditioning has been utilized to protect the heart from ischemia prior to ischemia onset, whereas postconditioning is employed to minimize the consequences of ischemia at the onset of reperfusion. The underlying mechanisms and pathways of ischemic pre- and postconditioning continue to be investigated as therapeutic targets. We evaluated the administration of a delta opioid agonist or cariporide on various parameters associated with myocardial reperfusion injury upon reperfusion of isolated porcine hearts. The hearts were reperfused in vitro with a Krebs buffer containing either: (1) 1 µM Deltorphin D (delta opioid specific agonist, n = 6); (2) 3 µM cariporide (sodium–hydrogen exchange inhibitor, n = 4); or (3) no treatment (control, n = 6). Subsequently, postischemic hemodynamic performance, arrhythmia burden, relative tissue perfusion, and development of necrosis were assessed over a 2 h reperfusion period. Postconditioning with Deltorphin D significantly improved diastolic relaxation (Tau, P < 0.05 versus controls) and decreased the incidence of ventricular arrhythmias during early reperfusion. Additionally, these treated hearts demonstrated increased tissue perfusion after 2 h (P < 0.05 versus controls), suggesting improved microvascular function. Delta opioid agonists elicited the potential to attenuate reperfusion injury, suggesting a postconditioning effect of these agents. We hypothesize that the induced benefits of delta opioids, in part, are associated with decreased calcium influx on reperfusion, independent of sodium–hydrogen exchange inhibition. Such agents may have a potential role in minimizing reperfusion injury associated with coronary stenting, bypass surgery, myocardial infarction, cardiac transplantation, or with the utilization of heart preservation systems.
Impact statement
In this study, we found that postconditioning with Deltorphin D significantly improved diastolic relaxation and decreased the incidence of ventricular arrhythmias during early reperfusion. Furthermore, these treated hearts demonstrated increased tissue perfusion after 2 h, suggesting improved microvascular function. Delta opioid agonists attenuated reperfusion injury, suggestive of a postconditioning effect. Such agents may have a potential role in minimizing reperfusion injury associated with coronary stenting, bypass surgery, myocardial infarction, cardiac transplantation, or with the utilization of heart preservation systems.
Introduction
Reperfusion injury is a paradox by which reperfusion, ultimately the most beneficial treatment after myocardial ischemia, causes additional injury to the myocardium. Characteristics of reperfusion injury after ischemia involve, but may not be limited to, myocardial stunning, arrhythmias, no-reflow phenomenon (microvascular damage), and accelerated cell death. 1 Furthermore, additional delayed injury may occur as molecular signals released from injured cells can induce apoptosis in bordering healthy cells. Importantly, reperfusion injury can potentially account for an estimated 25–60% of the total infarct volume following transient ischemia and reperfusion.2,3 Therefore, any potential reduction of reperfusion injury remains of great clinical interest, e.g. reperfusion injury after percutaneous coronary interventions, fibrinolysis, or after cardiac bypass surgery. Other relevant clinical scenarios may include cardiac transplantation and, most recently, assessment of ex vivo function prior to and as a bridge transplantation, for instance in TransMedic’s Organ Care™ System (Andover, MA, USA).4,5
Opioids have been reported to be beneficial when associated with either ischemic preconditioning 6 or pharmacological preconditioning, not only in animal models1,7–14 but also in humans.15,16 The induced effects included antiarrhythmic and anti-ischemic as well as cardioprotective benefits through various signaling mechanisms. The adaptation to ischemia observed in humans after two sequential coronary balloon inflations was abolished by naloxone, an opioid antagonist, 15 and the opioid receptor agonist DADLE was shown to precondition isolated human atrial trabeculae. 16 Moreover, opioids are commonly used in the intensive care unit for patients suffering from cardiac diseases requiring sedation, intubation, and cardiopulmonary resuscitation among other treatments. 17 While ischemic and opioid preconditioning have been convincingly shown to delay cell death in various experimental models, to date, the relative clinical applicability of the aforementioned observations has been limited to situations where the ischemic event can be anticipated. Yet, there is proven evidence that brief bouts of ischemia, subsequent to a prolonged ischemic event (ischemic postconditioning), can confer cardioprotection against reperfusion injury.18,19 Furthermore, pharmacological preconditioning agents (such as adenosine, bradykinin, and isoflurane) have also been shown to attenuate reperfusion injury when administered following ischemia. If pharmacological postconditioning could be shown to be beneficial for minimizing reperfusion injury, the administration of such agents could even be applied after restoration of blood flow. Opioid postconditioning has the potential to be developed into a safe and non-invasive therapeutic approach to reduce cardiac reperfusion injuries.20–26
It was initially assumed that cardioprotection would occur only when opioids were given prior to, but not during ischemia, i.e. as a cardioplegia additive.8,27 However, it has been subsequently shown that opioids attenuate reperfusion injury when administered upon reperfusion and even during a defined time frame up to 30 min afterward.11,26,28 Deltorphin D is a specific delta opioid receptor agonist.29,30 Significant myocardial infarct size reductions were observed when preconditioning swine with Deltorphin D at a dose of 1 mg/kg, compared with controls. 10
The overall aim of the present study was to investigate the ability of Deltorphin D to reduce reperfusion injury when administered as a supplement to the reperfusion buffer. Specifically, four parameters of reperfusion injury (myocardial stunning, arrhythmias, microvascular damage, and necrosis) were quantified. We employed an isolated working porcine heart model, which became subject to global ischemia; this model may mirror, to some degree, a transplant procedure or application of an ex vivo perfusion device. In addition, cariporide, a sodium–hydrogen exchange (NHE) blocker and an agent previously shown to attenuate reperfusion injury, was administered upon reperfusion and resulting effects were compared between therapies. 1
Materials and methods
This study was conducted in accordance with the Guide for the Care and Use of Laboratory Animals after approval from the Institutional Animal Care and Use Committee of the University of Minnesota.
Working isolated heart preparation
Briefly, swine (75–85 kg) were sedated with thiopental, then intubated and mechanically ventilated. Anesthesia was maintained using isoflurane and a 2:1 air-to-oxygen mixture. Medial sternotomies were performed exposing the hearts and major cardiac vessels. After cross-clamping of the aorta and great vessels, the hearts were arrested using cold (4℃) St. Thomas Hospital cardioplegia (110 mM NaCl, 16 mM KCl, 1.2 mM CaCl2, 16 mM MgCl2, 10 mM NaHCO3) administered under pressure (∼70 mmHg) via cannulas inserted into the ascending aortas. The hearts were excised and placed in a saline buffer slurry while the major vessels of the hearts were cannulated. Cardioplegia perfusion was maintained throughout the cannulation process to ensure the arrested state. The major vessels of the left ventricles (pulmonary veins and aortas) were fitted with clear tygon tubing cannulas and connected to the isolated heart apparatus. The hearts were then reperfused with warmed (37 ± 5℃), oxygenated (95% O2, 5% CO2) modified Krebs–Henseleit buffer (118 mM NaCl, 16 mM
Quantitative assessment of stunning and postischemic dysfunction
Hemodynamic and pressure data were collected prior to ischemia (in situ), at the initiation of working mode (Tw = 0 min), and for the subsequent 2 h, every 30 min. Left ventricular pressures were measured using Mikro-Tip catheter transducers (5 F, Model MPC-500, Millar Instruments, Inc., Houston, TX, USA). In vitro left atrial and aortic pressures were measured via pressure transducers placed at the inlets of the left atriums (pulmonary veins) and at the proximal ends of the descending aortas distal to the occluded brachiocephalic arteries, respectively. In addition, using 2 mm sonometric ultrasound crystals (Sonometrics, London, Ontario) placed on the endpoints of the two major axes of the left ventricles (four crystals total), time-dependent changes in left ventricle volume parameters were monitored (specifically, cardiac output and ejection fraction). Preload independent parameters of left ventricular end-diastolic pressure–volume relationship (EDPVR) and preload recruitable stroke work were measured by pressure–volume data collection during temporary occlusions of the inferior vena cavae or the pulmonary veins. All data were acquired with the Sonosoft program (Sonometrics) and postacquisition analyses were performed using the Cardiosoft program (Sonometrics).
Reperfusion arrhythmia assessment
Reperfusion arrhythmias were quantified during the first 45 min of working mode perfusion. A lead II electrocardiogram configuration was employed to monitor arrhythmias upon reperfusion in the isolated setting. Specifically electrodes were placed in the water bath proximal to the superior vena cava (Lead I), the left atrial appendage (Lead II), and the apex (Lead III). The electrocardiograms were amplified and filtered with a Spacelabs monitoring system (Model 1020, Spacelabs, Inc., Chatsworth, CA, USA) and analyses were completed using the Ponemah Physiology Platform Version 3.1 Program (Gould Instrument Systems, Inc., Valley View, OH, USA). The following modified scoring system was used to quantify arrhythmias: 0: <10 premature ventricular contractions (PVCs) in 9 min; 1: 10–50 PVCs in 9 min; 2: >50 PVCs in 9 min; 3: 1 episode of VF in 9 min; 4: 2–5 episodes of VF in 9 min; 5: >5 episodes of VF in 9 min.33,34
Microvascular damage and no-reflow assessment
For quantification of edema, tissue water contents were recorded prior to reperfusion as a baseline and again 2 h after working mode initiation by taking small (<100 mg) serial biopsies from the right atrial appendage (findings in our lab have indicated that serial left ventricular biopsies are associated with frequent arrhythmias). Additionally, regional myocardial blood flow was assessed with the use of two different colored 15 µm microspheres (1.0 × 106 per injection, E-Z TRAC Ultraspheres, Interactive Medical Technologies, Irvine, CA, USA) injected into the left atrium 15 min and 2 h after working mode start. After the study was completed, each left ventricle was frozen and cut into transverse 4 mm slices. Two transmural biopsies (∼3 g) were taken from the anterio-medial surface of the left ventricle. Each segment was separated into three layers (∼1 g each) for blood flow analysis: subendocardial, midmyocardial, and subepicardial. Routine tissue processing was completed (according to procedural instructions by Interactive Medical Technologies) and tissue perfusion was calculated using the formula
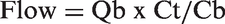
Metabolic status
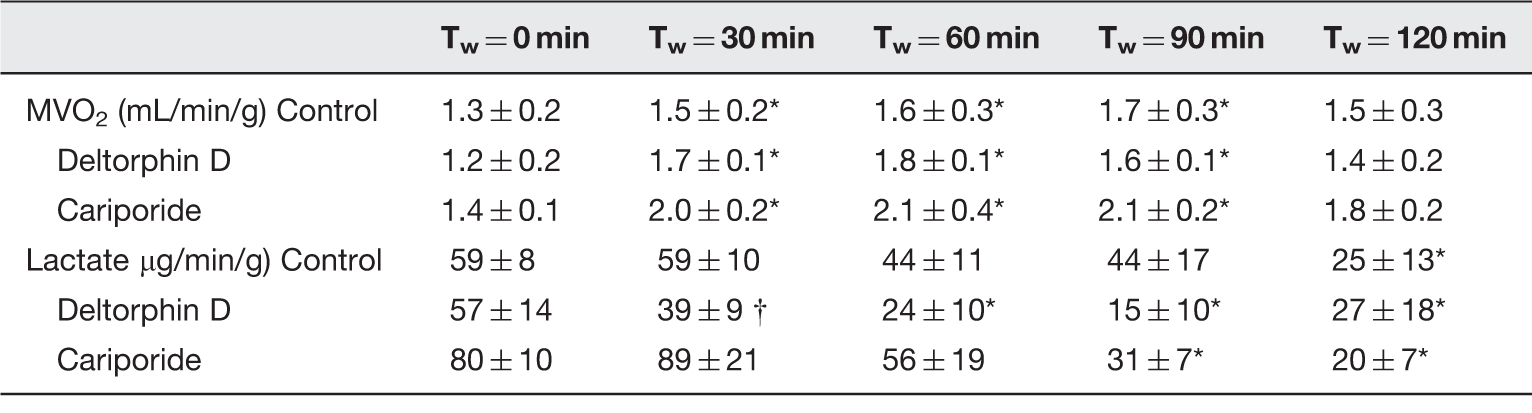
Serial 1 mL perfusate samples were simultaneously taken from the arterial (aorta) and venous coronary sinus (6 F Venaport coronary sinus guiding catheter, Cardima, Inc., Fremont, CA, USA) for metabolic analysis at working mode initiation (Tw = 0 min) and every 30 min thereafter. Blood gas measurements (pCO2, pO2, and pH; Instrumentation Laboratory 1640 BGE, Instrumentation Laboratory, Lexington, MA, USA) as well as lactate levels (Model 2300 Stat Plus Analyzer, Yellow Springs Instruments, Yellow Springs, OH, USA) were quantified and used for MVO2 and lactate efflux calculations. All data were normalized to prereperfusion heart weights (wet).
Necrosis assessment
Serial aliquots of perfusate (3 mL) were taken from the 6 F catheter placed in the coronary sinus to analyze creatine phosphokinase (CK) at working mode initiation (Tw = 0 min) and every 30 min thereafter. All values were normalized to prereperfusion heart weight (wet).
Protocol
Following 80 min of hypothermic (4℃) ischemia, the hearts were reperfused on the isolated heart apparatus (Figure 1). The reperfusion buffer was supplemented with the concentration listed below. The buffer was recirculated in the apparatus (through the coronary system) for 30 min in the Langendorff mode, after which a complete buffer change was accomplished and the hearts were made to function in working mode. Three groups of animals were used to study the effects of the delta opioid receptor agonist therapy administered on reperfusion; hearts were treated with either: (1) no reperfusion therapy (control, n = 6), (2) 1 µM Deltorphin D (n = 6), or (3) 3 µM cariporide (n = 4).
Experimental Protocol. Hemodynamic data were collected prior to ischemia, at working mode transition, and every 30 min thereafter. In treated hearts, Deltorphin D (1 µM) or cariporide (3 µM) was administered in the reperfusion buffer and circulated through the coronary system in Langendorff constant pressure retrograde perfusion for 30 min (stabilization period). Working mode transition occurred 30 min after reperfusion, just after opioid washout. Water content samples via right atrial biopsies were taken at baseline, prior to reperfusion, and 2 h postreperfusion. Arrhythmias were monitored during the first 45 min of working mode. Regional myocardial perfusion was assessed via colored microspheres 15 min (Tw = 15 min) after working mode transition and at Tw = 120 min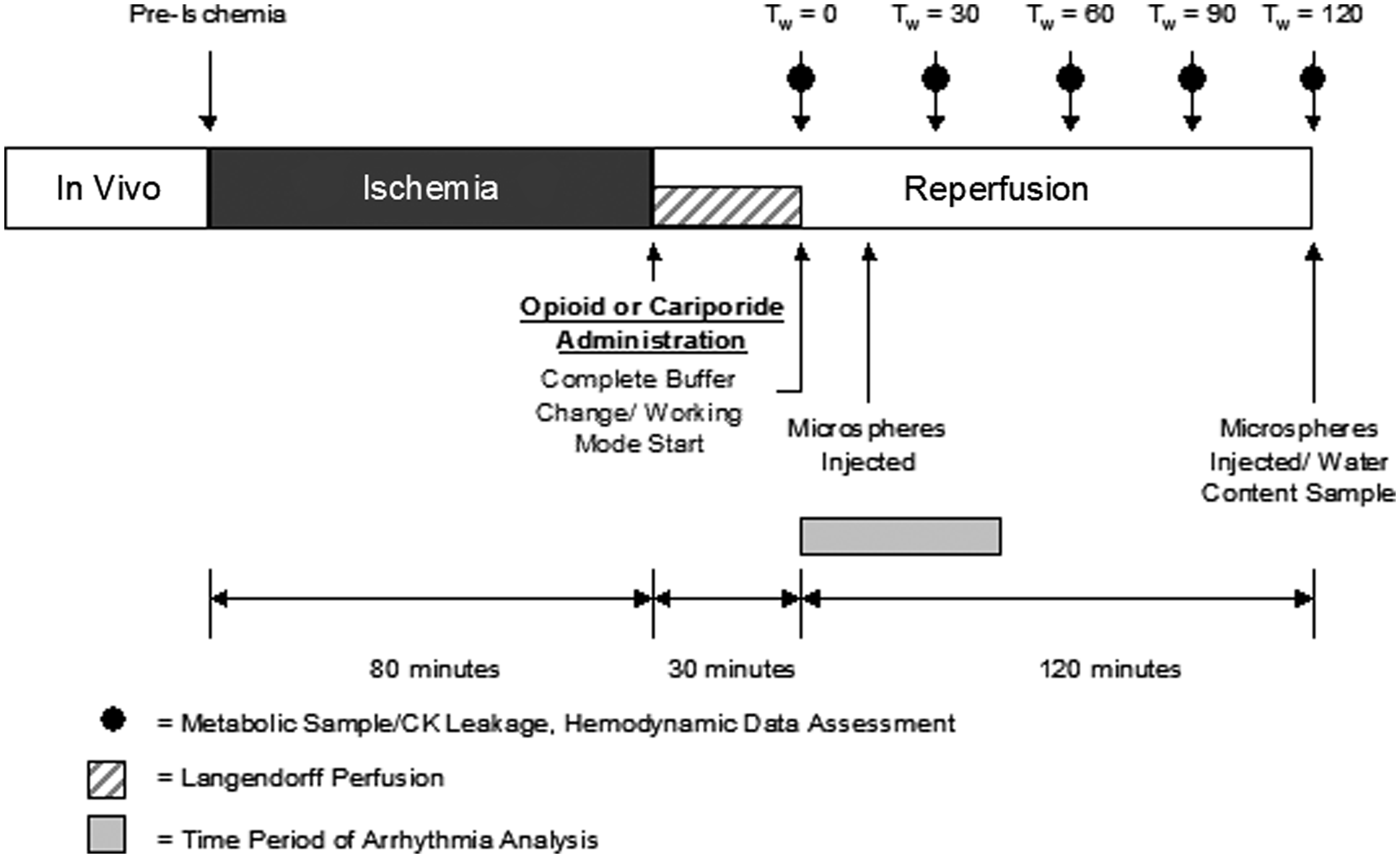
Statistical analysis
Data are reported as mean ± SEM. Data from all groups were analyzed using repeated measures ANOVA and Fisher’s protected least significant difference (PLSD) test as a post hoc test. One-way ANOVA analyses with Fisher’s PLSD were used to determine differences between groups at individual time points. All statistical analyses were completed using the Statview ® 5.0.1 program (SAS Institute Inc., Cary, NC, USA).
Results
Twenty-one experiments on isolated porcine hearts were attempted. Four of ten (40%) control hearts and one of seven (14%) Deltorphin D-treated hearts could not be transitioned into a sustained working mode due to insufficient left ventricular pressure development (<50 mmHg) and were subsequently excluded from the functional data analyses. Between groups of included animals, total body weights (controls: 81 ± 1 kg, Deltorphin D: 82 ± 6 kg, cariporide: 78 ± 2 kg) and wet heart weights (controls: 468 ± 34 g, Deltorphin D: 431 ± 26 g, cariporide: 442 ± 23 g) did not differ.
Postischemic dysfunction
Hemodynamic differences between groups were most pronounced immediately following working mode initiation (30 min postreperfusion, Tw = 0 min). Diastolic relaxation was significantly improved (as evidenced by a decreased diastolic relaxation time constant, Tau) in Deltorphin D-treated hearts compared to controls at Tw = 0 min (Figure 2). Left ventricular systolic pressure (LVSP) was decreased in the Deltorphin D-treated hearts at Tw = 0 min compared with controls (Table 1). Additionally, left ventricular contractility (dP/dtmax) was significantly lower relative to preischemic values at Tw = 0 min in the Deltorphin D group. Both control- and cariporide-treated hearts demonstrated decreased left ventricular diastolic compliance (increased left ventricular EDPVR) throughout the reperfusion period compared to baseline (preischemia) and relative to the Deltorphin D group (Table 1). Finally, lactate efflux as a measure of anaerobic metabolism declined significantly over the perfusion period with Deltorphin D treatment (Table 2).
Opioid effects on postischemic diastolic relaxation. Postischemic diastolic relaxation was impaired relative to baseline (preischemia) in all groups, as evidenced by significantly increased diastolic relaxation time constants (Tau). However, Tau was significantly lower in Deltorphin D-treated hearts during early reperfusion (Tw = 0 min) compared to controls, indicating improved relaxation with delta opioid treatment. *P < 0.05 versus preischemia (baseline); †P < 0.05 versus control Postischemic hemodynamic performance parameters All values are mean ± SE. Cariporide: 3 µM included in the reperfusion buffer; Tw = 0 min: working mode initiation (∼30 min postreperfusion and Deltorphin D administration); Control: no reperfusion therapy; Deltorphin D: 1 µM Deltorphin D included in the reperfusion buffer. CO: cardiac output (in L/min); LV dP/dtmax: maximal first derivative of left ventricular pressure (mmHg/s); LVEDP: left ventricular end-diastolic pressure (in mmHg); LVSP: left ventricular systolic pressure (in mmHg); LV EDPVR: stiffness constant (kp) of left ventricular end-diastolic pressure–volume relationship (mL−1); PRSW: slope of preload recruitable stroke work of the left ventricle (in mmHg/mL). P < 0.05 versus preischemia (repeated measures ANOVA); †P < 0.05 versus control (ANOVA). Postischemic metabolic parameters All values are mean ± SE. Cariporide: 3 µM included in the reperfusion buffer; Control: no reperfusion therapy; Deltorphin D: 1 µM Deltorphin D included in the reperfusion buffer; Tw = 0 min, working mode initiation (∼30 min postreperfusion and Deltorphin D administration); Lactate: lactate efflux across the heart (in µg/min per gram heart weight); MVO2: myocardial oxygen consumption (in mL O2/ min per gram heart weight). P < 0.05 versus Tw = 0 min (repeated measures ANOVA); †P < 0.05 versus cariporide (ANOVA).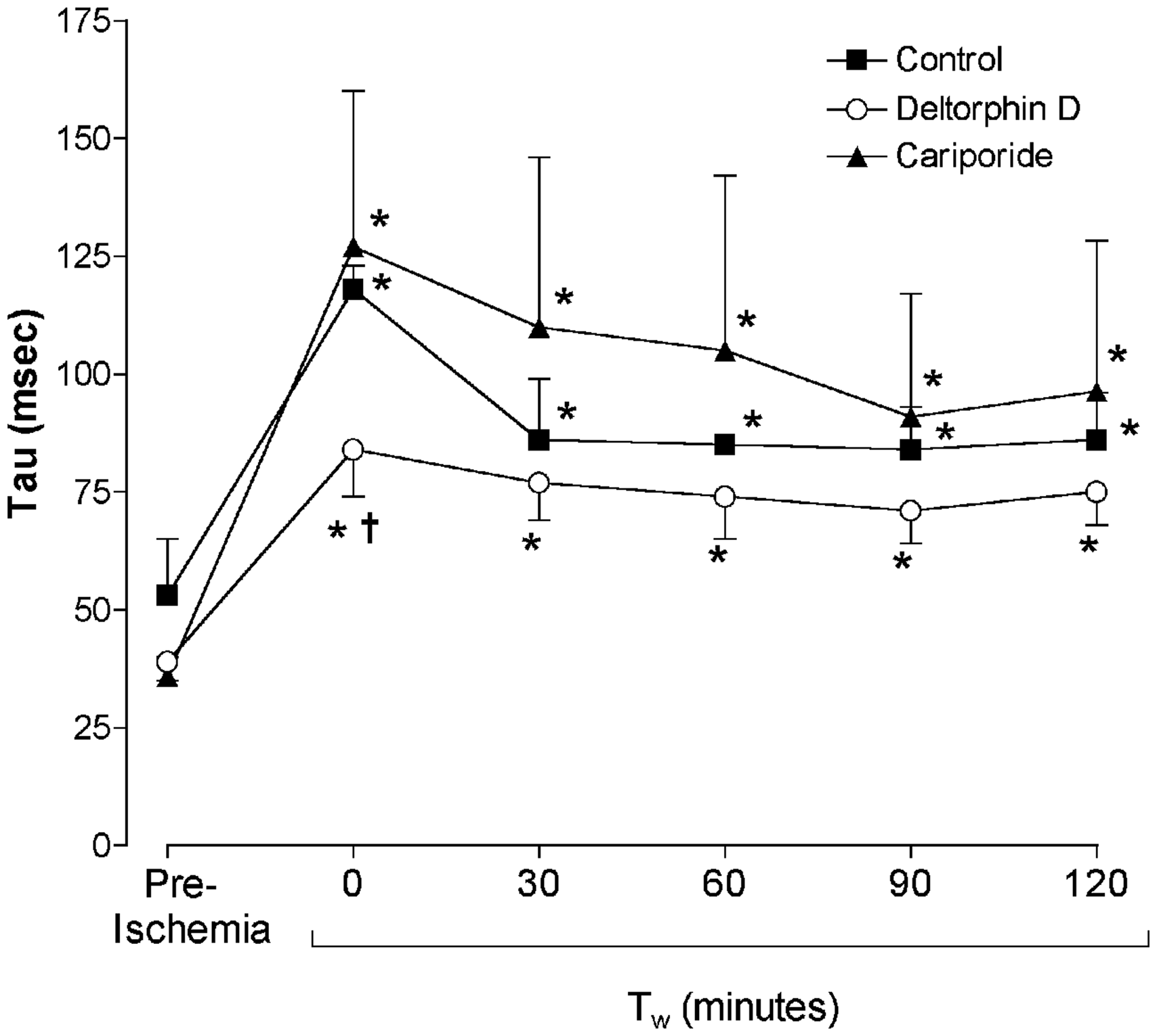

Reperfusion arrhythmias
All hearts elicited VF upon reperfusion and required internal defibrillation. There were no significant differences between groups in the number of defibrillatory shocks needed to convert to normal sinus rhythm. As displayed in Figure 3, compared to control and cariporide, Deltorphin D-treated hearts presented with fewer arrhythmias during the first 9 min of working mode and, therefore, showed decreased reperfusion arrhythmia scores (Control: 1.3 ± 0.2 versus Deltorphin D: 0.8 ± 0.2, P < 0.1). Cariporide-treated hearts demonstrated a significantly increased incidence of PVCs and consequently higher arrhythmia scores compared with Deltorphin D during the second arrhythmia scoring period (10–18 min; P < 0.05).
Opioid effects on ventricular arrhythmias during reperfusion. Shown is a comparison of ventricular arrhythmia scores during the first 45 min of working mode perfusion. Quantification of arrhythmias using a scoring system (scored every 9 min) showed a decrease in relative arrhythmia occurrence with Deltorphin D during the early periods of working mode perfusion compared to the cariporide group. While mean arrhythmia scores were lower in the Deltorphin D-treated hearts compared to controls during the first 9 min of working mode, this difference was not significant (P = 0.1). *P < 0.05 versus cariporide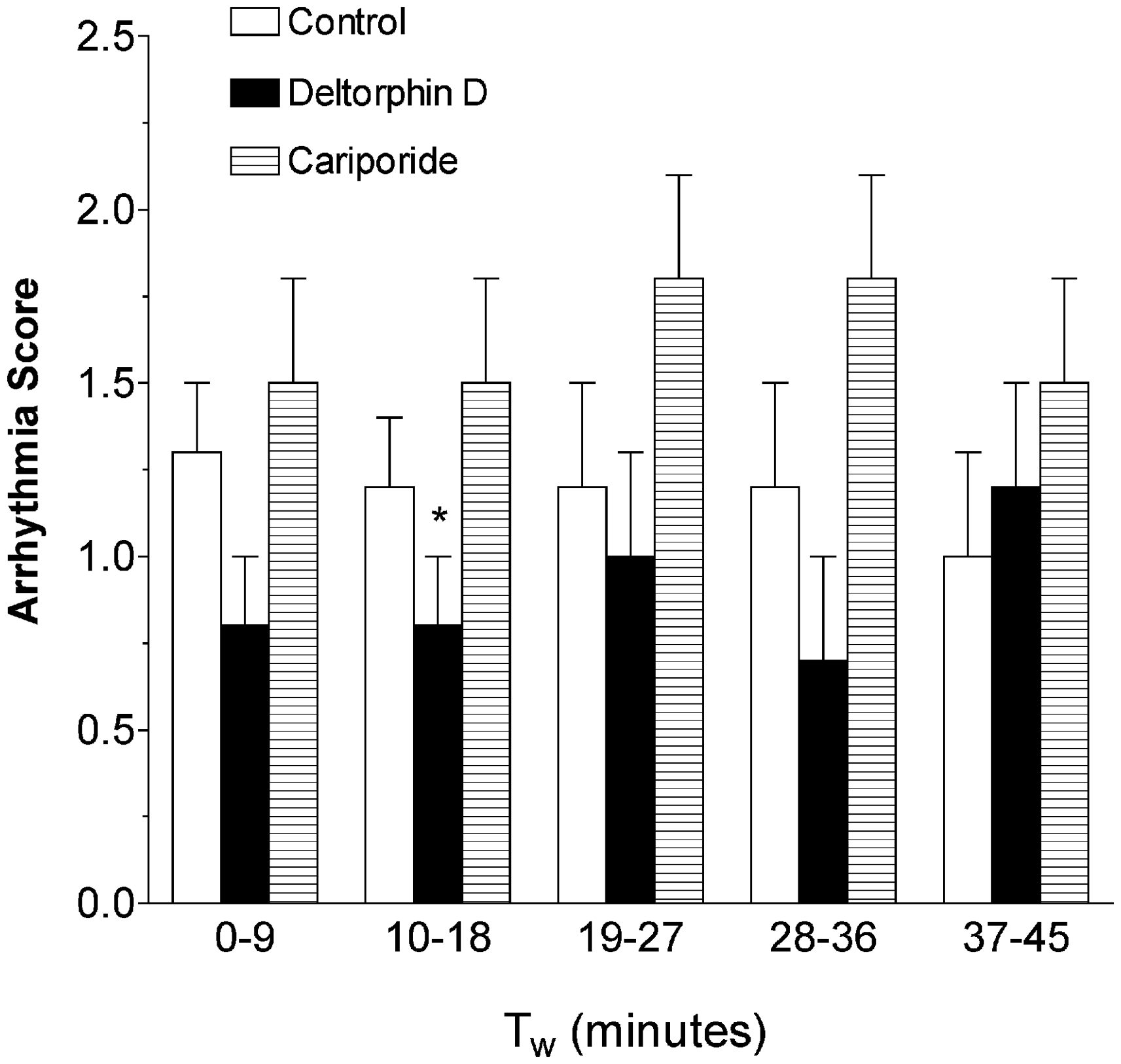
Microvascular damage
Edema development (i.e. water content) at Tw = 120 min was not statistically different between groups (%H2O: control = 66 ± 3%; Deltorphin D = 57 ± 6%; cariporide =67 ± 4%). Tissue perfusion data collected over the reperfusion period via serial colored microsphere injections are shown in Figure 4. Comparisons were only made between control- and Deltorphin D-treated hearts (n = 4 each), as these data were not collected in cariporide-treated hearts. Control hearts showed minimal change in perfusion of the subendocardial, midmyocardial, and subepicardial layers over the perfusion period. Conversely, Deltorphin D-treated hearts showed increased tissue perfusion compared to controls at Tw = 120 min, which was significant in the midmyocardial layer (P < 0.05).
Opioid effects on postischemic myocardial tissue perfusion. Regional myocardial perfusion of the anterior left ventricular wall was assessed using colored microsphere injections at 15 and 120 min after working mode initiation (Tw = 0). Transmural biopsies were taken from the myocardium at experiment termination and separated into three layers: (a) subendocardial, (b) midmyocardial, and (c) subepicardial. Flows were normalized per gram of tissue. Preliminary evidence of the no-reflow phenomenon was seen at the Tw = 120 time point, as all control hearts showed declining trends in flow to all layers of the heart. This trend was not present in the Deltorphin D-treated hearts and a significant increase in flow at Tw = 120 was found in the midmyocardial level. *P < 0.05 versus control
Necrosis
At the initiation of working mode (Tw = 0 min) there was a trend of increased creatine kinase activity in the perfusate from controls; however, there was no significant difference between groups (control: 0.16 ± 0.13 versus Deltorphin D: 0.08 ± 0.08 IU/L/g; Figure 5). Additionally, hearts in both groups elicited similar levels of creatine kinase activity in the perfusate during the first hour of reperfusion and thereafter. Creatine kinase activity in the perfusate collected from cariporide-treated hearts was not detectable with spectrophotometric analysis (<20 IU/L/g).
Opioid effects on reperfusion-induced necrosis. Perfusate creatine kinase (CK) activity expressed per gram of heart was used to quantify reperfusion-induced necrosis. Perfusate CK activity in the cariporide-treated hearts was undetectable. There was no significant reduction in necrosis with Deltorphin D administration compared to controls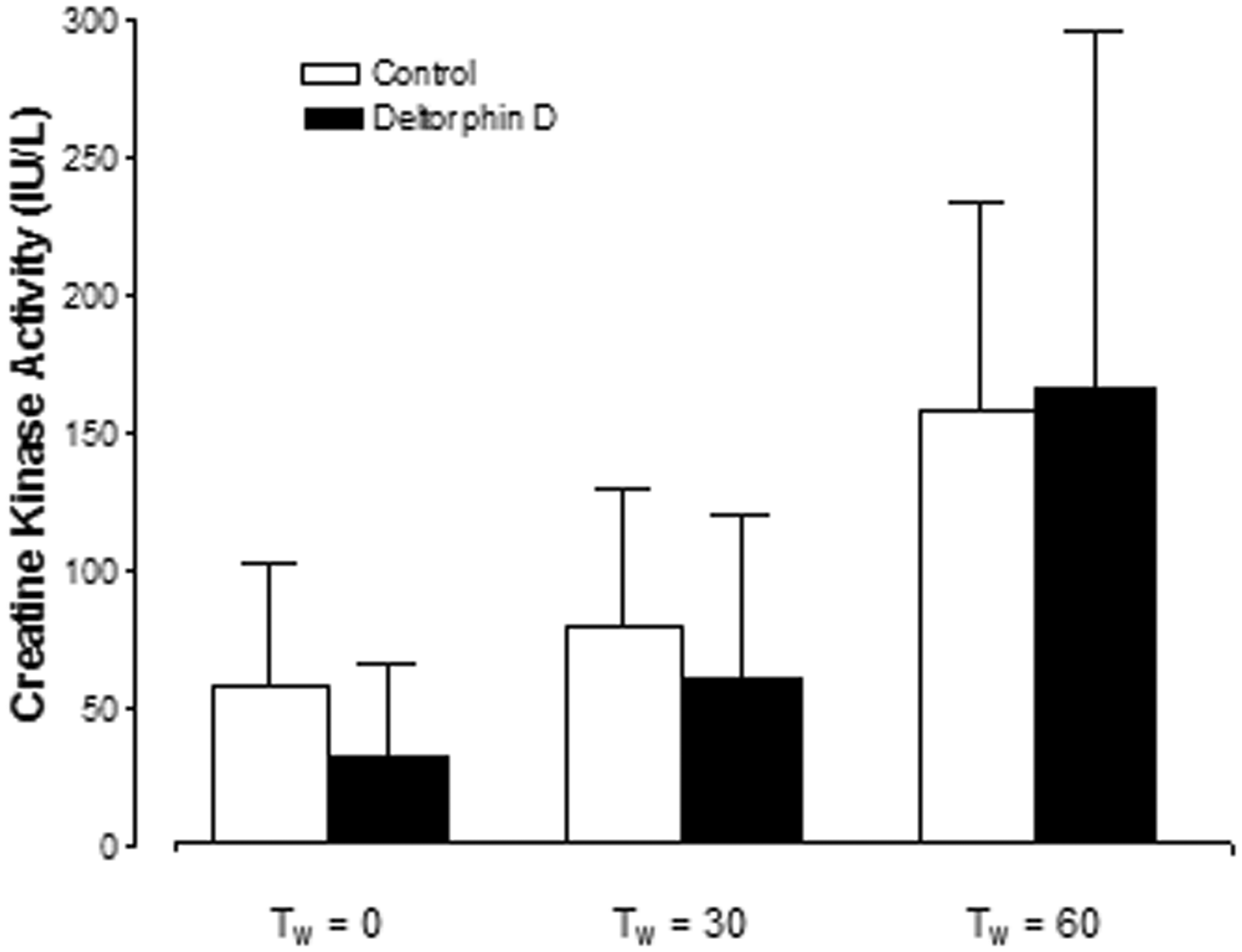
Discussion
In the present study, we demonstrated that the postischemic administration of Deltorphin D, a specific delta 2 opioid agonist, during the initial reperfusion period significantly attenuated subsequent reperfusion injury in isolated working porcine hearts. While Deltorphin D supplemented to the reperfusion buffer initially transiently decreased systolic LVSP and contractility (dP/dtmax) compared to control hearts, subsequently there were no differences in systolic performance between the experimental groups. Deltorphin D treatment was associated with enhanced left ventricular diastolic compliance (left ventricular EDPVR) and relaxation (Tau). Furthermore, Deltorphin D also prevented decreases in tissue perfusion (no-reflow phenomenon) over time and decreased reperfusion-induced arrhythmias. The administration of cariporide, a NHE blocker, upon reperfusion failed to demonstrate any functional benefits and increased the incidence of reperfusion arrhythmias. Interestingly, necrosis determination showed no detection of CK, which could indicate less necrosis development in comparison to the Deltorphin D and control groups. This finding might offset cariporide’s aforementioned negative effects during reperfusion. Nevertheless, these results collectively show, for the first time, that delta opioid postconditioning is effective in attenuating dysfunction during the early reperfusion period.
As in the current study, similar protocols with other known pharmacological preconditioning agents such as adenosine, bradykinin, and isoflurane have been shown to mitigate reperfusion injury. For example, the selective A1/A2 agonist AMP 579 has been proved to attenuate reperfusion injury (reduce infarct size) in swine when administered either prior to ischemia (preconditioning) or during the early reperfusion period. 35 Similarly, in the globally ischemic and reperfused isolated rabbit hearts, administration of AMP 579 at reperfusion decreased infarct size and reduced postischemic contracture. 22 In addition to those studies, Ke et al. 36 found that postconditioning with adenosine also inhibited inflammation by dropping myocardial NF-κB and TNF-α expression significantly in the rat myocardium. Furthermore, administration of different mu opioid receptor selective drugs (e.g. remifentanil, fentanyl, butorphanol, etc.) has elicited attenuations in reperfusion injury as postconditioning agents.11,14,37–39 Lastly, it was reported that the addition of DADLE, a delta and mu opioid agonist, to the reperfusion buffer of a postischemic isolated rat heart prevented the development of ventricular arrhythmias. However, it should be noted that specific blockade of the mu opioid receptor abolished this effect. 40
Previous work from our laboratory demonstrated that preconditioning with the delta 2-specific agonist, Deltorphin D, significantly decreased infarct size in a porcine coronary occlusion model while kappa opioid receptor co-activation during preconditioning exacerbated the ischemic insult.10,41 In addition to its infarct limiting ability, Deltorphin D preconditioning was also associated with decreased LVSP and contractility (dP/dtmax) during ischemia and early reperfusion compared with controls. Similar to our preconditioning studies, in the present study postconditioning with Deltorphin D decreased systolic performance (LVSP and dP/dtmax) compared to controls during the early reperfusion period. Of additional interest, isolated rat hearts preconditioned with DPDPE, a delta 1-specific opioid agonist, also demonstrated decreased reperfusion-induced ventricular arrhythmias with contractile dysfunction (negative inotropic effect) upon reperfusion. 42
As was found with adenosine, 43 Deltorphin D administration attenuated the no-reflow phenomenon exhibiting a significant increase in coronary flow in the midmyocardial layer at the 2 h reperfusion time point. The no-reflow phenomenon is a multifactorial syndrome that is potentially caused by leukocyte-mediated endothelial damage; however, it may also present as a secondary consequence of myocyte edema and contracture.1,44 In the leukocyte-free crystalloid perfused heart employed in the present study, we found early evidence of the no-reflow phenomenon in control hearts only (i.e. trends of decreased myocardial perfusion over time). The Deltorphin D post-treated hearts also elicited trends for decreased edema development and significantly improved diastolic performance during the reperfusion period, possibly implicating attenuation of microvascular damage via this secondary mechanism.
While we did not find a decrease in necrosis (creatine kinase efflux) with Deltorphin D administration during reperfusion compared to controls, it should be noted that the severity of the ischemia stimulus is considerably less in a hypothermic globally ischemic model compared to a normothermic coronary occlusion model. Also of interest, in a previous study from our laboratory using the same isolated porcine model of global ischemia, we did not find a reduction in necrosis but rather improved hemodynamic performance with morphine or DADLE preconditioning of isolated porcine hearts. 32 It should be noted that reperfusion injury is a multifaceted phenomenon and lack of protection against reperfusion-induced necrosis does not necessarily preclude that reperfusion injury was not attenuated.
The underlying mechanisms of ischemic postconditioning are considered to be multifactorial and involve cell-surface receptor activation, survival signaling pathways, and mitochondria as the end effectors. Calcium is known to play an important role in the pathophysiology of reperfusion injury, as reperfusion-induced calcium overload is one of the primary instigators of myocardial stunning, reperfusion arrhythmias, and necrosis.1,29,38,45–48 The data collected in this study lead us to speculate that opioid administration on reperfusion may alter sarcolemmal or mitochondrial calcium fluxes. In an additional attempt to elucidate the role of sarcolemmal calcium influx on reperfusion injury in our isolated heart model, we administered the NHE blocker, cariporide, during the reperfusion period. It has been shown in previous studies that cariporide secondarily prevents calcium overload by attenuating sodium influx and subsequently preventing sarcolemmal calcium overload upon reperfusion. 49 Many investigators have demonstrated that administration of NHE blockers prior to ischemia limits the extent of ischemia–reperfusion injury. However, when this therapy was administered during the early reperfusion period, there were conflicting opinions as to the benefits. While we found similar trends of decreased LVSP with either cariporide or Deltorphin D administration, which may be due to decreased intracellular calcium during systole, there were no marked benefits of cariporide treatment. Furthermore, cariporide administration in this model produced a pro-arrhythmogenic effect during the early reperfusion period compared to Deltorphin D and controls (Figure 3). Hence, it may be speculated that in this isolated heart model, decreased sarcolemmal calcium influx alone does not have a significant effect on reperfusion injury.
It is generally accepted that endogenous opioid ligands are involved in the process of ischemic pre- and postconditioning, making the opioid receptor system a logical drug target to achieve similar effects. The opioid receptor is composed of seven transmembrane domains and belongs to the class A family of G protein-coupled receptors. These are involved in cardiac stress signaling and cardioprotection by activation of PI3K/Akt and RISK pathway components. 14
Delta opioid-induced cardioprotection during reperfusion may be due, in part, to opening of sarcolemmal and mitochondrial KATP channels, subsequently preventing mitochondrial Ca2+ overload during reperfusion and thus preserving postischemic mitochondrial energy production (i.e. decreased lactate production) and enhancing diastolic relaxation.11,50
The no-flow phenomenon was lessened in patients when the KATP channel opener, nicorandil, a coronary vasodilator and pharmacological preconditioning agent, was administered as an adjunct to reperfusion therapy. 51 Similarly, results from the IONA Study Group52 and CESAR 53 clinical trials demonstrated that nicorandil, when given to patients with unstable angina, decreased cardiovascular events associated with ischemia, including a decreased incidence of arrhythmias. Murata et al. 54 reported that opening of the MPT during reperfusion, secondary to cytosolic and mitochondrial calcium overload, may be primarily responsible for reperfusion-induced cellular death. They also showed that MPT opening is a reperfusion-specific phenomenon that does not occur during ischemia and that diazoxide, a mitochondrial KATP channel opener, attenuates mitochondrial Ca2+ loading and, consequently, MPT opening during reperfusion. 54 Collectively, these results lead us to assume that opioid-induced cardioprotection during reperfusion may be due partly to opening of mitochondrial KATP channels, and subsequently preventing mitochondrial Ca2+ overload during reperfusion, thus preserving postischemic mitochondrial energy production (i.e. decreased lactate production) and enhancing diastolic relaxation. Furthermore, decreased calcium influx with Deltorphin D treatment would also explain the observed decrease in systolic performance during the early reperfusion period found in both the preconditioning and present studies.
Despite the fact that Deltorphin D administration did not completely ameliorate ischemia–reperfusion injury in the present study, altering the dose and timing of administration may prove more successful. The dose employed in our study was chosen based on preliminary preconditioning studies in an acute coronary occlusion model 10 and in vitro isolated muscle bath preparations (unpublished data). Whereas benefits were observed in these models, due to the novel design of this study, direct extrapolation of these results to administration upon reperfusion was not possible. Additionally, since the Tw = 0 min data collection time point appeared to show the most functional benefit of opioid administration, it may be of greater benefit to increase the timing of administration on reperfusion.
This pharmacological approach has its clinical application as an addition to thrombolysis medication and during coronary angioplasty procedures. In this setting, the drug could be administered early and allow time to actually circulate through the body until onset of coronary reflow. 29 Also myocardial ischemia and reperfusion injury both negatively affect the outcome of cardiac transplantation. With advances in normothermic ex vivo organ perfusion, pharmacological postconditioning is gaining a new dimension and importance. The discovery of drugs that effectively attenuate reperfusion injury and better maintain cardiac function on the device, or even induce functional recovery, is relevant.
In this study, we utilized a large mammalian porcine model, in comparison to small animals and rodents that were mostly used in the past for postconditioning studies.37,55 The pig serves as an appropriate acute surgical, transplantation, and ischemia model presenting with similar anatomic and physiologic characteristics to humans for this experimental setup, making this research translational.56,57 We conclude that the investigation of this specific delta opioid as a postconditioning agent in the porcine model may confer clinical relevance in minimizing reperfusion injury associated with coronary stenting or bypass surgery, myocardial infarction, or cardiac transplantation. Further additional reperfusion studies on heart preservation systems utilizing the specific delta opioid would be revealing.
Footnotes
Authors’ contributions
MS assisted with data analyses and manuscript preparation. JAC and DCS developed the experimental protocol, collected data, and supported the manuscript preparation. PAI guided the experimental design, performed surgical procedures, assisted with data collection, edited the manuscript, and secured funding for the research.
Acknowledgements
We would like to thank Peter Oeltgen for his advice and providing us with Deltorphin D. We acknowledge Alexander Hill and Anna Legried Dopp for their feedback, and Monica Mahre for assistance with manuscript submission. This research was supported by the Lillehei Heart Institute and the Institute for Engineering in Medicine at the University of Minnesota and by Medtronic (Minneapolis, MN, USA).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.