
Abstract
Mutations in genes encoding nuclear lamins and associated nuclear envelope proteins have been linked to a broad range of inherited diseases affecting different tissues and organs. These diseases are often referred to as laminopathies. Scientists have yet to elucidate exactly how pathogenic mutations leading to alteration of a nuclear envelope protein cause disease. Our relatively recent research has shown that pathogenic mutations in genes encoding nuclear envelope proteins lead to defective nucleocytoplasmic connections that disrupt proper functioning of the linker of nucleoskeleton and cytoskeleton complex in the establishment of cell polarity. These defects may explain, at least in part, pathogenic mechanisms underlying laminopathies.
Impact statement
Mutations in genes encoding nuclear lamins and associated nuclear envelope proteins have been linked to several diseases affecting different tissues and organs. The pathogenic mechanisms underlying these diseases, often called laminopathies, remain poorly understood. Increased knowledge of the functions of different nuclear envelope proteins and the interactions between them is crucial to elucidate these disease mechanisms. Our research has shown that pathogenic mutations in genes encoding nuclear envelope proteins lead to defective nucleocytoplasmic connections that disrupt proper functioning of the linker of nucleoskeleton and cytoskeleton (LINC) complex in the establishment of cell polarity. These defects may contribute to the pathogenesis of laminopathies and provide novel targets for therapeutics.
Keywords
Introduction
Genetics research has since the middle of the 1990s linked mutations in genes encoding proteins of the nuclear envelope to rare diseases. Most of these diseases involve specific tissues and organs even though the affected proteins are fairly ubiquitously expressed. One notable example is mutations in LMNA encoding lamin A and lamin C, components of the nuclear lamina that are expressed in most terminally differentiated cells. Mutations in LMNA can cause myopathy, cardiomyopathy, partial lipodystrophy, peripheral neuropathy and multi-system disorders, such as progeria, which have features of accelerated aging. Diseases caused by mutations in LMNA and genes encoding B-type lamins and other nuclear envelope proteins, most of which are directly or indirectly associated with lamins, are often referred to as “laminopathies.” 1
The nuclear envelope separates the nucleoplasm from the cytoplasm in nucleated eukaryotic cells. In addition to its barrier function, recent data have emerged showing that the nuclear envelope connects the nuclear lamina inside the nucleus to the cytoskeleton in the cytoplasm. The linker of nucleoskeleton and cytoskeleton (LINC) complex mediates these connections. Several laminopathy-causing mutations disrupt LINC complex-mediated nucleocytoplasmic connections, leading to various cellular defects. These defects may relate to disease pathogenesis.
To better understand of potential pathogenic mechanisms underlying the laminopathies, one must first understand basic aspects of the nuclear envelope and the diseases themselves. We have therefore structured this review to first discuss the composition and molecular genetics of the nuclear envelope, with an emphasis on the lamins and LINC complex proteins. We then discuss genetic and clinical aspects of different laminopathies. Finally, we address the potential role of defective nucleocytoplasmic connections in these diseases.
Nuclear envelope and nucleocytoplasmic connections
The nuclear envelope is composed of the nuclear pore complexes, nuclear lamina, and nuclear membranes, which are continuous with the rough endoplasmic reticulum (Figure 1). The pore complexes provide routes for passive and active transport between the nucleus and cytoplasm and also function in chromatin organization and regulation of gene expression.2,3 The lamina, which lines the inner aspect of the inner nuclear membrane, is composed of intermediate filament proteins called lamins.4–7 Electron microscopy of Xenopus oocytes originally identified the nuclear lamina as meshwork of filaments 10 nm in diameter, which is similar to cytoplasmic intermediate filaments. 8 More recent cryo-electron tomographic analysis of mouse embryonic fibroblasts lacking vimentin revealed that lamin filaments have a globular-decorated fiber appearance of 3.5 nm thickness. 9 Hence, in mammalian somatic cells, the lamina may have a distinctly different structure than canonical cytoskeletal intermediate filament networks.

Schematic diagram of the nuclear envelope showing nuclear pore complexes, the nuclear lamina and the nuclear membranes. The nuclear membranes are morphologically divided into the pore membrane, inner nuclear membrane, and outer nuclear membrane. The outer nuclear membrane is directly continuous with the rough endoplasmic reticulum (ER) membrane and similarly contains ribosomes (represented by small black circles within these membranes). (A color version of this figure is available in the online journal.)
Nuclear membranes are morphologically divided into the pore, inner and outer membrane domains. The pore membranes are small domains of the nuclear envelope that connect the inner and outer membrane and contain transmembrane proteins of the nuclear pore complexes such as gp210 and POM121.10,11 The inner nuclear membrane is associated with the nuclear lamina. Approximately 80 transmembrane proteins have been identified as specifically targeted to the inner nuclear membrane in interphase rat hepatocytes 12 ; however, the transmembrane protein composition of the inner nuclear membrane may vary between cell types. 13 Most of these transmembrane proteins are synthesized on rough endoplasmic reticulum-bound ribosomes and reach the inner nuclear membrane by lateral diffusion through the interconnected rough endoplasmic reticulum, outer nuclear, and pore membrane domains. They are then retained in the inner nuclear membrane by binding to the lamina or chromatin. During this diffusion, the nucleocytoplasmic domains of the proteins pass through the 10 nm diameter lateral channels of nuclear pore complexes, which prevents proteins with large nucleocytoplasmic domains from reaching the inner nuclear membrane this way.14–17 However, mechanisms involving active transport have been postulated as alternatives to this diffusion and binding process. 18
The outer nuclear membrane has ribosomes on its outer surface and is separated from the inner nuclear membrane by the perinuclear space, a continuation of the endoplasmic reticulum lumen. As there is no barrier for diffusion between the rough endoplasmic reticulum and the directly continuous outer nuclear membrane, both have long been presumed to have identical integral membrane protein content. However, interactions between the luminal domains of transmembrane proteins in the inner nuclear membrane and luminal domains of transmembrane proteins of the rough endoplasmic reticulum could lead to specific retention of proteins in the outer nuclear membrane. This is what happens at the LINC complexes that connect the nucleus and cytoskeleton. 19
LINC complexes are composed of outer nuclear membrane KASH (Klarsicht, ANC1, and Syne homology) proteins and inner nuclear membrane SUN (Sad1 and UNC-84) proteins, both of which are type II membrane proteins with a single transmembrane segment. In mammals, somatic cell KASH proteins are generally referred to as nesprins. 20 Nesprins contain a KASH domain of approximately 60 amino acids at their carboxyl-termini, which includes a transmembrane segment and up to 30 residues in the perinuclear space. They also contain multiple, clustered spectrin repeats that project into the cytoplasm. SUN proteins contain a conserved SUN domain also located within the perinuclear space. X-ray crystallography analysis shows that three KASH peptides bind to a trimeric arrangement of the SUN domains of SUN2, with a KASH-SUN disulfide bond adding stability to the complex.21,22 The other widely expressed SUN protein, SUN1, may exist in a different oligomeric state. 23
Binding of SUN proteins to nesprins leads to formation of a protein “bridge” between the inner and outer nuclear membranes (Figure 2). Inside the nucleus, SUN proteins interact with the lamina, specifically A-type lamins.19,24 In mammalian cells lacking A-type lamins, SUN proteins still localize to the inner nuclear membrane. This suggests that associations with other nuclear proteins or perhaps chromatin contribute to their localization. However, SUNs and nesprins demonstrate increased diffusional mobility within the nuclear membrane in cells lacking A-type lamins, suggesting that these proteins contribute to stabilizing LINC complexes in the nuclear envelope. 25 SUN proteins also interact with emerin and SAMP1, integral proteins of the inner nuclear membrane.26,27 On the cytoplasmic side of the nuclear envelope, LINC complexes, via their different nesprin components, bind to cytoskeletal elements. Different nesprin proteins (see below) bind to different cytoskeletal elements via their variable cytoplasmic domains. Various nesprin-1 and nesprin-2 isoforms interact with the microtubule motors dynein or kinesin-1.27–29 Nesprin-1G and nesprin-2G bind to actin filaments via calponin homology domains near the aminotermini.30,31 Nesprin-2G can additionally bind to actin filaments via the formin FHOD1 and the actin bundling protein fascin.32,33 Nesprin-3 binds to plectin, which in turn interacts with cytoplasmic intermediate filaments. 34 Nesprin-4 interacts with kinesin-1. 35 These nesprin-mediated connections to different cytoskeletal elements allow LINC complexes to function in diverse cellular processes.

Schematic diagram of LINC complexes. Transmembrane SUN proteins of the inner nuclear membrane (INM) interact with nesprins of the outer nuclear membrane (ONM) within the perinuclear space. In the nucleus, SUN proteins bind to A-type lamins and also interact with emerin, another transmembrane protein of the INM. In the cytoplasm, different nesprin proteins bind to different cytoskeletal elements. Different isoforms of nesprin-1 and nepsrin-2 can bind actin filaments directly or associate with microtubules indirectly via dynein or kinesin-1. Nesprin-3 can associate with intermediate filaments by binding to plectin. Nesprin-4 can also associate with microtubules indirectly by binding kinesin-1. (A color version of this figure is available in the online journal.)
Molecular genetics of the lamina and LINC complex
In mammals, three genes encode lamins, the protein building blocks of the nuclear lamina. The proteins are generally divided into two types, A-type and B-type, based on their biochemical properties and primary structures. 36 LMNB1 on human chromosome 5 encodes lamin B1, which is expressed in all or most somatic cells.37,38 LMNB2 on human chromosome 19 encodes lamin B2, also expressed in all or most somatic cells. 39 In mammals, the gene encoding lamin B2 also gives rise to a male germ cell‐specific isoform, lamin B3, which is generated by alternative RNA splicing. 40
LMNA on human chromosome 1 is the human gene encoding the A-type lamins, prelamin A and lamin C, which arise by alternative RNA splicing and are expressed in most terminally differentiated cells.38,41 Prelamin A and lamin C are identical for their first 566 amino acids with prelamin A having 98 unique carboxyl-terminal amino acids and lamin C having 6. In mammals, the same gene that encodes lamin A and C also gives rise to a germ cell‐specific isoform, lamin C2, and a poorly studied minor isoform called lamin AΔ10.42,43
Except for lamin C, mammalian lamins terminate with a CaaX motif, where C is a cysteine, a an aliphatic amino acid, and X any amino acid. This motif at the carboxyl-termini of proteins triggers three sequential enzymatic modifications. First, protein farnesyltransferase catalyzes the addition of a farnesyl moiety to the cysteine. 44 Second, an endoprotease that recognizes the farnesylated protein catalyzes cleavage of the peptide bond between the cysteine and -aaX. For B-type lamins and most other CaaX proteins, ras converting CAAX endopeptidase 1 (RCE1) catalyzes this reaction. 45 For prelamin A, zinc metallopeptidase, STE24 homolog (ZMPSTE24) can also catalyze the cleavage of the -aaX. 46 In the third step, isoprenylcysteine carboxyl methyltransferase catalyzes methylation of the carboxyl-terminal farnesylcysteine.47,48
In the case of prelamin A, but not B-type lamins, the protein undergoes an additional reaction to generate mature lamin A. This is an endoproteolytic cleavage leading to removal of its last 15 amino acids, which includes the farnesylcysteine α-methyl ester.49,50 Indeed, the cysteine must be farnesylated for this cleavage reaction to occur. 50 Experiments in both knockout mice and in vitro have shown that ZMPSTE24 catalyzes this endoproteolytic cleavage reaction.46,51,52 The physiological reason for this processing of prelamin A to lamin A remains unknown and the cleaved 15-amino-acid polypeptide has never been detected in cells. However, defects in this processing lead to human disease (see below).
The mammalian genome contains five genes that encode somatic cell KASH-domain proteins, four of which are called nesprins. The human gene symbols for the four nesprins are SYNE1 through SYNE4, for synaptic nuclear envelope, because the encoded proteins were originally identified in the nuclei that lie beneath the postsynaptic membrane at the neuromuscular junction. 53 The proteins were subsequently named nesprins for nuclear envelope spectrin-repeat proteins. 20 SYNE1 and SYNE2 encode nesprin-1 and nesprin-2, respectively, with multiple isoforms of each arising by alternative RNA splicing and alternative translational start and termination sites.20,54 SYNE1 contains 146 exons and SYNE2 116 exons and the “giant” or “G” isoforms are >800 kDa. Some smaller isoforms reside in the inner nuclear membrane, where they associate with emerin, SUN proteins, and lamins.26,55 SYNE3 encodes nesprin-3, which also has several splice variants and is expressed widely in somatic cells. 34 Nesprin-4, encoded by SYNE4, is absent from many tissues and preferentially expressed in secretory epithelia and hair cells of the inner ear.35,56 A fifth human gene, LRMP, encodes Jaw1/LRMP, which contains a KASH domain and interacts with SUN proteins in lymphoid organs, immune cells, taste buds, and melanomas. 57 CCDC155 encodes KASH5, which is expressed only in germ cells and associates with telomeres, SUN1, and dynactin during meiosis.58,59
In mammals, five different genes code for SUN proteins. SUN1 and SUN2 encode proteins that are widely expressed in in many cell types.60,61 SUN3, SPAG4, and SUN5 encode testis-specific SUN-domain proteins.62–64
Laminopathies
Laminopathies are rare, inherited diseases caused by mutations in genes encoding nuclear lamins and other nuclear envelope proteins. Here, we focus on those laminopathies affecting A-type nuclear lamins, the SUN and nesprin core proteins of the LINC complex and the LINC complex-associated protein emerin. More details on these diseases and the laminopathies involving other nuclear envelope proteins can be found in our previous review articles.1,65–70
A-type nuclear lamins
Different mutations in LMNA can generate four major, mostly non-overlapping phenotypes selectively involving striated muscle, adipose tissue or peripheral nerve or involving multiple systems with progeroid features (Figure 3). The first striated muscle disease linked to mutations in LMNA was autosomal dominant Emery-Dreifuss muscular dystrophy, which is characterized by dilated cardiomyopathy and a scapulohumeral-peroneal distribution of skeletal muscle involvement with concurrent tendon contractures. 71 Subsequent research showed that the same dominant LMNA mutations that cause amino acid substitutions, small deletions, splicing defects or haploinsufficiency can cause related disorders with dilated cardiomyopathy but variable muscle involvement.72–75 Extremely rare compound heterozygous mutations can cause the same striated muscle phenotypes. 76 Dominant mutations that mostly cluster in exon 8 of LMNA cause Dunnigan-type familial partial lipodystrophy, in which there is selective loss of subcutaneous fat from the extremities, excessive fat accumulation in the neck and face, and the subsequent development of insulin resistance, diabetes mellitus, and liver steatosis.77–79 The vast majority of these mutations cause amino acid substitutions that change the surface charge of an immunoglobulin-type fold in lamin A and lamin C.80,81 However, variant lipodystrophy syndromes and metabolic disorders have been described caused by mutations affecting amino acids in different domains of the proteins.82,83 Homozygosity for the R298C LMNA mutation causes Charcot–Marie–Tooth type 2B1, a sensory and motor peripheral neuropathy with chronic distal weakness and often associated with foot and spine deformations. 84 This very rare disorder has only been identified in Northwest African families whose affected individuals share a common ancestral haplotype. 85 Finally, specific mutations in LMNA cause progeroid syndromes, which involve multiple organ systems and in some respects mimic premature aging.

LMNA mutations cause four main disease phenotypes. Most dominant LMNA mutations cause striated muscle disease. The diagram shows the classical Emery-Dreifuss muscular dystrophy, which is characterized by dilated cardiomyopathy and a scapulohumeral-peroneal distribution of skeletal muscle involvement with concurrent tendon contractures. However, the same mutations can result in dilated cardiomyopathy with variable skeletal muscle involvement. Other dominant mutations most often leading to a change in the surface charge of the immunoglobulin fold in lamins A and C cause Dunnigan-type partial lipodystrophy, which has selective loss of subcutaneous fat from the extremities, excessive fat accumulation in the neck and face with the subsequent development of insulin resistance, diabetes mellitus, and liver steatosis. The autosomal recessive R298C LMNA mutation causes a Charcot–Marie–Tooth type 2 peripheral neuropathy characterized by a stocking-glove sensory neuropathy, an associated pes cavus foot deformity and additional variable features such as scoliosis. The de novo dominant G608G LMNA mutation causes the multisystem disease HGPS, with progeroid features including growth retardation, micrognathia, reduced subcutaneous fat, alopecia, osteoporosis, skin mottling, and premature vascular occlusive disease. Other rare dominant LMNA mutations also cause progeriod syndromes with similar features; the recessive R527H mutation causes mandibuloacral dysplasia, a disorder with a combination of progeroid features and partial lipodystrophy. Reprinted from Dauer and Worman 66 with permission from Elsevier. (A color version of this figure is available in the online journal.)
Hutchinson-Gilford progeria syndrome (HGPS) is the prototypical progeroid syndrome. Characteristic features are growth retardation, micrognathia, reduced subcutaneous fat, alopecia, osteoporosis, skin mottling and premature vascular occlusive disease, with affected children dying in their teenage years usually from complications of cardiovascular disease. 86 It is caused by a de novo G608G (ggc > ggt) or G608S (ggc > agc) mutation in LMNA.87,88 These mutations lead to the creation of an aberrant splice donor site in a portion of RNA encoded by exon 11, which leads to an in-frame deletion of 50 amino acids in prelamin A. This internally truncated prelamin A, which is called progerin, retains the farnesylated and methylated carboxyl-terminal cysteine of prelamin A. Considerable evidence points to progerin, specifically farnesylated progerin, as being the major driver of pathology in HGPS. Accumulation of progerin in cells leads to alterations in nuclear architecture. 89 Treatment of cultured cells expressing progerin with a protein farnesyltransferase inhibitor (FTI), which blocks protein farnesylation, reverses the abnormal nuclear architecture.90–95 More significantly, FTI treatment improves the phenotype of genetically-modified mice that express progerin.96,97 An FTI has since been studied in clinical trials of children with HGPS.98,99
Deletion of Zmpste24 leads to expression of full-length but unprocessed, permanently farnesylated prelamin A and progeroid phenotypes in mice.51,52 FTI treatment significantly improves the progeroid phenotype in these mice, demonstrating that the farnesyl moiety of the unprocessed prelamin A contributes to pathology. 100 In humans, mutations in ZMPSTE24 cause a range of progeroid disorders from the relatively mild mandibuloacral dysplasia type B to a severe progeria syndrome to the neonatal lethal restrictive dermopathy.101–105 ZMPSTE24 has other functions than the proteolytic processing of prelamin A106 but a correlation between residual proteolytic activity in mutant forms of ZMPSTE24 and the severity of the progeroid disorders they cause further emphasizes the role of unprocessed prelamin A in these diseases. 105 We have also described one patient with a progeroid disorder and a point mutation that abolishes the ZMPSTE24 recognition site in prelamin A. Cultured dermal fibroblasts from this individual have abnormal nuclear morphology that is reversed when treated with a FTI. 107
The evidence is strong that protein farnesylation contributes to pathology in most of the progeroid disorders associated with prelamin A. However, there are several cases of mutations in LMNA causing progeroid disorders with similar features without evidence of accumulation of a farnesylated prelamin A variant. Mandibuloacral dysplasia type A has features of both progeria and lipodystrophy and is caused by a recessive LMNA R527H mutation. 108 Cases classified as atypical Werner syndrome have also been associated with LMNA mutations and farnesylated prelamin A variants similarly do not appear to be expressed. 109 Hence, rare mutations in LMNA leading to alterations in A-type lamins other than those causing accumulation of farnesylated prelamin A variants can also cause progeroid phenotypes.
SUN and nesprin core proteins of the LINC complex
Various diseases have been linked to mutations in the genes encoding SUN proteins and nesprins, some robustly and some more tentatively. 110 Mutations in SYNE1 encoding nesprin-1 cause autosomal recessive cerebellar ataxia. 111 Since the original publication, subsequent studies have confirmed that mutations in SYNE1 may be one of the more common recessive ataxias worldwide.112,113
SYNE1 mutations also cause recessive arthrogryposis multiplex congenita, a disorder characterized by congenital joint contractures and reduced fetal movements, and the genetic linkage is robust with pathogenic alleles shown to segregate with affected individuals in different families.114–116 Homozygosity for a mutation in SYNE4, which encodes a truncated nesprin-4 variant, also segregates with affected individuals in two families of Iraqi-Jewish ancestry with progressive high-frequency hearing loss. 56 An autosomal dominantly inherited point mutation in SYNE2 leading to an amino acid substitution in nesprin-2β1 segregates among first-degree relatives with an Emery-Dreifuss muscular dystrophy-like phenotype. 117
Other mutations in genes encoding SUN proteins and nesprins have been linked to cardiomyopathy and muscular dystrophy. Autosomal dominant sequence variations in SYNE1 have been reported in individuals with cardiomyopathy, sometimes associated with Emery-Dreifuss muscular dystrophy-like skeletal muscle involvement.117–119 Sequence variations in SUN1 and SUN2 have also been reported in individuals with phenotypes similar to Emery-Dreifuss muscular. 120 However, these SYNE1, SUN1, and SUN2 sequence variants have not been shown to segregate between affected and unaffected individuals within families, making these associations with cardiomyopathy and muscular dystrophy tentative.
LINC complex-associated protein emerin
Positional cloning in families with X-linked Emery-Dreifuss muscular dystrophy identified EMD encoding emerin as the causative gene. 121 Subsequently, emerin was localized to the inner nuclear membrane and shown to be lacking from the nuclear envelope of affected patients.122,123 Almost all cases are associated with loss of emerin expression.122–125 The phenotype of X-linked Emery-Dreifuss muscular dystrophy is virtually identical to that of the autosomal form caused by LMNA mutations, with dilated cardiomyopathy as a predominant feature. Mutations in EMD causing lack of emerin expression can also cause dilated cardiomyopathy with different patterns of skeletal muscle involvement.126,127
Defective nucleocytoplasmic connections in laminopathies
To gain novel insights into pathogenesis, our groups have been investigating how pathogenic mutations that cause laminopathies affect nucleocytoplasmic connections. We have used a modified wounded fibroblast monolayer system to examine the initial polarization of cells preparing for migration.128,129 Addition of serum or the serum factor lysophosphatidic acid (LPA) to serum-starved, wounded monolayers triggers rapid polarization of the wound-edge cells, which is characterized by reorientation of the centrosome between the nucleus and the wound. Centrosome reorientation in wounded monolayers of fibroblasts does not involve movement of the centrosome but rather movement of the nucleus past a stationary centrosome by myosin-directed retrograde actin flow. 130
LPA stimulation of wounded fibroblast monolayers activates two pathways, one that induces this nuclear movement and a second that holds the centrosome in place (Figure 4). LPA stimulation leads to activation of Cdc42. 131 In the nuclear movement pathway, Cdc42 activates myotonic dystrophy kinase-related Cdc42-binding kinase, which phosphorylates and activates myosin II. 130 This leads to retrograde actin cable flow originated from the wound edge. The nucleus is coupled to these moving actin filaments via the LINC complex, specifically nesprin-2G-SUN2 which binds to actin via the calponin homology domains of nesprin. Linear arrays of nesprin-2G and SUN2 assemble to form transmembrane actin-associated nuclear (TAN) lines to move the nucleus rearward. 31 Cdc42 simultaneously activates a second pathway involving Par6/PKCζ, Par3, dynein/dynactin, and microtubules that holds the centrosome in the cell centroid.130,132 These pathways acting together lead to proper cell polarity. Defects in either of these pathways lead to different cellular phenotypes. If the nuclear movement pathway is blocked, both the centrosome and nucleus are positioned at the cell centroid. If the centrosome centration pathway is blocked, both are positioned rearward of the cell centroid.
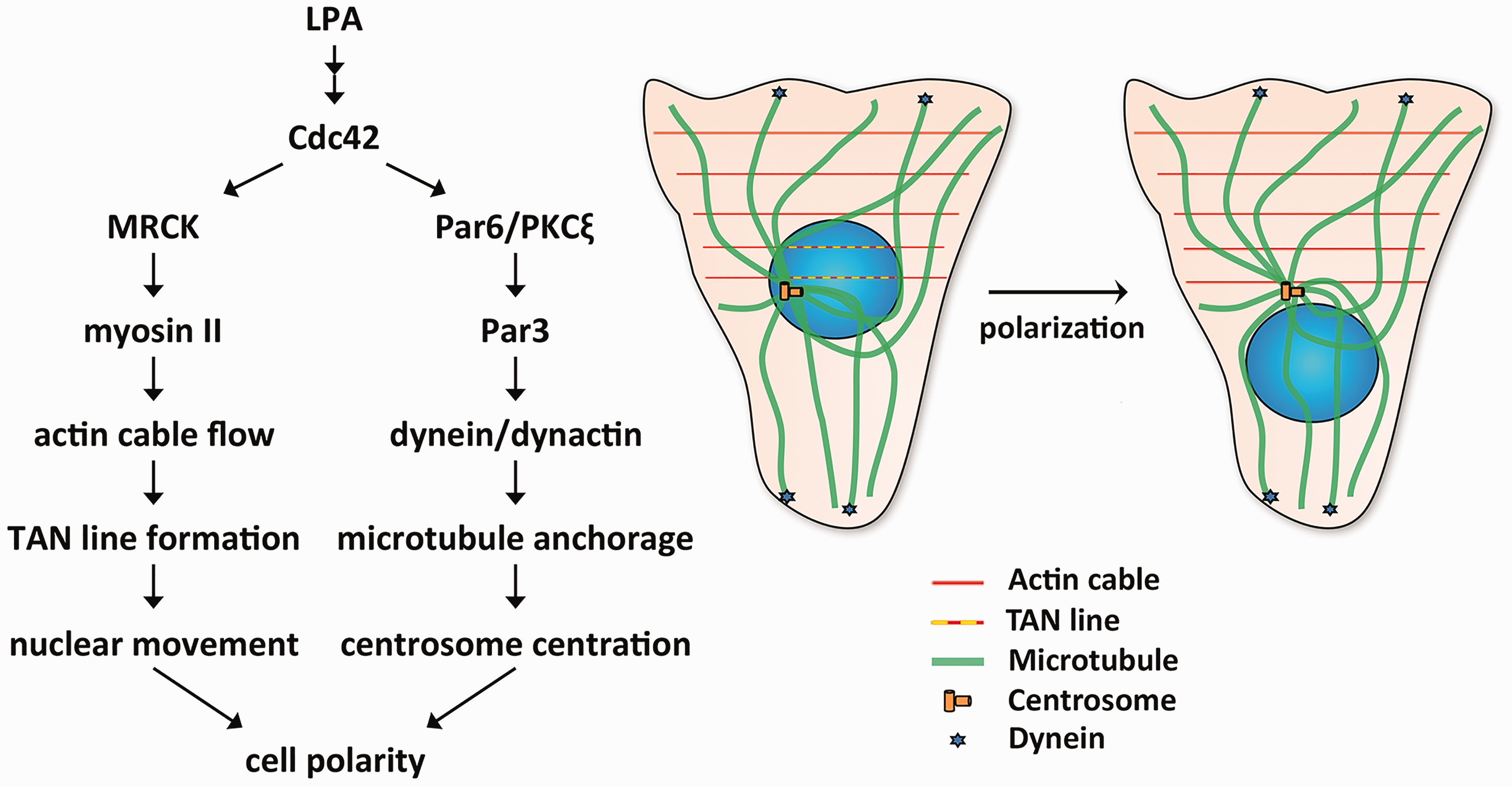
Mechanisms that generate cell polarity in wounded monolayers of fibroblasts. LPA-treatment of wounded monolayers leads to activation of Cdc42. Cdc42 activates two pathways that lead to cell polarity. In the nuclear movement pathway (left arm of diagram at left), Cdc42 activates myotonic dystrophy kinase-related Cdc42-binding kinase (MRCK), which phosphorylates and activates myosin II, leading to actin cable flow and TAN line formation to move the nucleus. Cdc42 simultaneously activates a second centrosome centration pathway (right arm in the diagram at left), which involves Par6/PKCζ, Par3, dynein/dynactin, and microtubules and holds the centrosome in the centroid of the cell. The schematic diagram at the right shows microtubules, actin cables, TAN lines, and dynein during cell polarization. (A color version of this figure is available in the online journal.)
Striated muscle disease
We initially examined how lamin A variants expressed in either striated muscle disease or Dunnigan-type partial lipodystrophy affect fibroblasts in wounded monolayers polarizing for migration. 133 Expression of lamin A variants found in patients with striated muscle diseases blocked actin-dependent nuclear movement, whereas most of the partial lipodystrophy variants inhibited microtubule-dependent centrosome centration. Depletion of A-type lamins similarly blocked nuclear movement, showing that lamin A variants affecting striated muscle generate a null phenotype. This is consistent with in vivo findings that germline deletion of A-type lamins in mice leads to striated muscle disease but not lipodystrophy.134,135 TAN lines assembled within cells expressing striated muscle disease-associated lamin A variants or depleted of A-type lamins; however, they appeared less stable and slipped over the nucleus rather than moving with it. Hence, alterations in A-type lamins that occur in striated muscle disease cause defective anchoring of TAN lines and inhibit cell polarization by blocking actin-dependent nuclear movement.
In subsequent experiments, we examined how loss of the LINC complex-associated integral membrane protein emerin, which occurs in X-linked Emery-Dreifuss muscular dystrophy, affects fibroblasts polarizing for migration. 136 In fibroblasts without emerin, TAN lines formed normally but nuclei moved randomly due to nondirectional actin cable flow. Slippage of TAN lines was also observed. Depletion of myosin IIB from cells generated similar nondirectional nuclear movement. Emerin interacts with myosin IIB and is required for myosin IIB to localize near the nucleus. These results are consistent with a model in which emerin is necessary for the proper organization of cytoplasmic actin flow through localization of myosin IIB. Consistent with its role, emerin is not uniquely localized to the inner nuclear membrane but a small quantity is normally in the outer nuclear membrane. 137
Although the mechanisms may be different, genetic alterations that occur in striated muscle diseases caused by LMNA and EMD mutations both prevent effective establishment of polarity in wounded monolayers of fibroblasts polarizing for migration. SUN1 and SUN2 variants tentatively linked to striated muscle disease also block nuclear movement and cause polarity defects in the same assay. 120 However, defects in fibroblast polarity and subsequent migration toward a wound edge cannot be obviously linked to the striated muscle pathology in these diseases. Yet, establishing polarity in migrating myoblasts is essential for skeletal muscle development and repair. In experiments using wounded monolayers of cultured myoblasts, we established that essentially the same processes are involved in the establishment of polarity as in fibroblasts, including stimulation by LPA and rearward movement of the nucleus while the centrosome is maintained at the cell centroid. 138 As in fibroblasts, nuclear movement was dependent on actin, the formation of nesprin-2G and SUN2 TAN lines and the presence of A-type lamins. Furthermore, abolishing polarization by depleting nesprin-2G interfered with directed myoblast migration and fusion into myotubes. Defects in actin-dependent nuclear movement in myoblasts, a process first identified in fibroblasts, may therefore play a role in the pathogenesis of striated muscle diseases caused by mutations in LMNA and EMD. Still, myoblast dysfunction cannot explain all of the pathology, as mice with germline deletions of A-type lamins, emerin or even both proteins are born with skeletal muscle, which in the case of emerin loss alone is only minimally abnormal well into adulthood.134,139–141 Furthermore, affected humans usually do not develop symptoms until later childhood or early adulthood. A plausible mechanism, which remains to be tested experimentally, would be that alterations in A-type lamins or emerin also make differentiated myofibers more susceptible to mechanical damage and that partially defective myoblasts cannot adequately replace the damaged fibers.
HGPS
More recently, we have examined nucleocytoplasmic connections in HGPS. 142 LPA-stimulated wounded monolayers of fibroblasts form children with HGPS, as well as NIH3T3 fibroblasts expressing progerin, demonstrated defective nuclear movement and polarity establishment. These defects were reversed by blocking progerin farnesylation. Farnesylated progerin inhibited rearward nuclear movement by both weakening TAN line anchorage, as occurs in cells lacking A-type lamins, and disturbing retrograde actin flow.
SUN1 over accumulates in cells from children with HGPS.26,143 We showed that progerin expression increases SUN1 accumulation in NIH3T3 fibroblasts and SUN1 overexpression prevents rearward nuclear movement and polarization in wounded monolayers. 142 Furthermore, SUN1 depletion by siRNA rescues the defects in rearward nuclear movement and polarization in fibroblasts from children with HGPS. SUN1 accumulation inhibits nuclear movement in these cells by inducing excessive association of microtubules with nuclei. This agrees with our previous finding that SUN1 is involved in microtubule-dependent nuclear positioning. 29 Consistently, inhibition of dynein rescues actin-dependent nuclear movement in fibroblasts from children with HGPS and NIH3T3 cells overexpressing progerin. Hence, imbalanced nucleocytoplasmic connections resulting from increased SUN1 expression and the resultant preferential engagement of microtubules over actin by LINC complexes underlie the polarity defects in fibroblasts from children with HGPS. Intriguingly, fibroblasts from human subjects older than approximately 60 years exhibit similar nuclear movement and cell polarity defects as fibroblasts from children with HGPS and these defects can be rescued by reducing SUN1 levels or inhibiting dynein. 142
Experiments in mouse models further suggest that elevated level of SUN1 in progeroid disorder leads to physiological defects. Progeroid mice with the LmnaΔ9 mutation express a truncated, farnesylated prelamin A, albeit different than progerin, and live approximately 30 days. 144 However, genetic deletion of Sun1 in these mice improves growth and prolongs survival. 143 Mice with an Lmna point mutation corresponding to that causing HGPS express abundant amounts of progerin and have a progeroid phenotype, including a distinctive aortic pathology with loss of smooth muscle cells in the arterial media and fibrosis of the adventitia. Expression of a dominant-negative KASH-domain construct that disrupts the LINC complex in smooth muscle cells ameliorates the toxic effects of progerin in these cells and limits the accompanying adventitial fibrosis. 145 These results suggest that modulating SUN1 expression or disrupting LINC complexes in progeroid disorders in which there are excessive connections to microtubules could be effective therapies in children with HGPS and even the cardiovascular complications of physiological aging.
Conclusions
Genetic mutations causing laminopathies can generate defective nucleocytoplasmic connections. These in turn lead to nuclear positioning and cell polarity defects. Proper nuclear positioning and establishing and maintaining polarity are necessary for productive cell migration and the proper development and function of diverse organs.146–148 The relatively recent data generated from studies of defective nucleocytoplasmic connections in laminopathies can therefore lead to compelling hypotheses about pathogenesis for further experimental testing. These may explain, at least in part, pathogenic mechanisms underlying diseases caused by mutations in genes encoding nuclear envelope proteins.
Footnotes
Authors’ contributions
All of the authors contributed to this review.
ACKNOWLEDGMENTS
The authors thank the Guest Editor of this Thematic Issue for the invitation to contribute this review.
DECLARATION OF CONFLICTING INTERESTS
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: HJW declares the following potential conflicts of interest: scientific advisory board and equity owner AlloMek Therapeutics, scientific advisory board and consulting income MNG Laboratories, consulting income Eiger BioPharmaceuticals, sponsored research funding (to Columbia University) Navitor Pharmaceuticals. The other authors declare no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
FUNDING
The author(s) disclose receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health [grant numbers R01AR048997, R01AR068636].