
Abstract
Fetal Alcohol Spectrum Disorders (FASDs) describe a range of deficits, affecting physical, mental, cognitive, and behavioral function, arising from prenatal alcohol exposure. FASD causes widespread white matter abnormalities, with significant alterations of tracts in the cerebral cortex, cerebellum, and hippocampus. These brain regions present with white-matter volume reductions, particularly at the midline. Neural pathways herein are guided primarily by three guidance cue families: Semaphorin/Neuropilin, Netrin/DCC, and Slit/Robo. These guidance cue/receptor pairs attract and repulse axons and ensure that they reach the proper target to make functional connections. In several cases, these signals cooperate with each other and/or additional molecular partners. Effects of alcohol on guidance cue mechanisms and their associated effectors include inhibition of growth cone response to repellant cues as well as changes in gene expression. Relevant to the corpus callosum, specifically, developmental alcohol exposure alters GABAergic and glutamatergic cell populations and glial cells that serve as guidepost cells for callosal axons. In many cases, deficits seen in FASD mirror aberrancies in guidance cue/receptor signaling. We present evidence for the need for further study on how prenatal alcohol exposure affects the formation of neural connections which may underlie disrupted functional connectivity in FASD.
Impact statement
Despite public health campaigns to deter drinking during pregnancy, maternal alcohol consumption remains a major public health concern. This review describes axon guidance mechanisms that are vulnerable to developmental alcohol exposure. Additionally, it highlights the importance of investigating the effects of alcohol exposure early in gestation which has been under-studied. This review provides a framework for understanding the range of pathologies in FASD. Furthermore, we provide specific future directions for research in the cerebral cortex, cerebellum, and hippocampus, regions of the brain that are well established targets of developmental alcohol exposure.
Introduction
Fetal alcohol spectrum disorder (FASD) is an umbrella term used to describe the range of physical, mental, and behavioral deficits that present in an individual exposed to alcohol in the womb. 1 Three brain regions that are highly vulnerable to prenatal alcohol exposure are the cerebral cortex, cerebellum, and hippocampus. In the cerebral cortex, the corpus callosum (CC) connects the two hemispheres; proper connection here is necessary for processing, decision-making, and responding to incoming information. The cerebellum coordinates voluntary movement, posture, balance, and motor learning. It also plays a role in cognitive functions and emotional control. The hippocampus, responsible for learning and memory, is one of the most alcohol-sensitive brain regions because of its dynamic responsiveness to the extracellular environment. Gross brain abnormalities such as reduced volume and defects in functional connectivity in these brain regions manifest in FASD.1,2 Several guidance mechanisms underlying the development of the cerebral cortex, cerebellum, and hippocampus are shared. Axons, more specifically their highly dynamic tips known as growth cones, are directed by guidance cue/receptor families, guidepost cells, and axon scaffolds along stereotypical trajectories to their target destinations. Aberrant axon guidance mimics some cellular, molecular, and behavioral phenotypes of FASD, but few studies focus on the impact of alcohol toxicity on these mechanisms. In this review, we focus on guidance cue/receptor families that are critical to form major tracts in the cerebral cortex, cerebellum, and hippocampus: Semaphorin/Neuropilin, Netrin/DCC, and Slit/Robo. 3 We highlight tracts in each region—the CC in the cerebral cortex, climbing and mossy fibers in the cerebellum, and mossy fibers in the hippocampus. We aim to explore FASD pathology potentially associated with axon guidance in these regions.
Cerebral cortex—Corpus callosum
Guidepost cell populations initiate callosal axon guidance across the midline
Proper connection between the hemispheres requires axon guidance by guidepost cells, axon scaffolds, and guidance molecules. 4 Emigrant glutamatergic and GABAergic neuron populations serve as guidepost cells, traveling to the midline before pioneer axons from the cingulate cortex begin to approach and cross, followed by callosal axons forming the CC. The guidepost neurons are transient, only residing at the midline during the embryonic stages. 5 Glutamatergic neurons, thought to migrate from the retrobulbar ventricle, arrive first and form the subcallosal sling. 5 These are followed by GABAergic neurons, which originate in the medial ganglionic eminence (MGE). 5 Glial cells that form the glial wedge and indusium griseum also serve as guidepost cells at the midline, secreting guidance cues to guide pioneer axons from the cingulate and callosal axons. 6
Semaphorin signaling attracts both cingulate and callosal axons to the midline
Semaphorin signaling in the midline mediating formation of the CC is regulated by ephrin type-B receptor 1 (EphB1), nuclear factor 1 B-type (Nfib) transcription factor, and neuropilin-1 (NRP1) receptor.5,7,8 In addition to the glutamatergic neurons that serve as guideposts, Sema3C is expressed by another cell population at the midline, cells of the subcallosal sling.6,9 Glutamatergic neurons are concentrated at the midline and the subcallosal sling forms a ventral boundary below the CC. 10 Midline Sema3C expression first attracts pioneering axons 9 originating in the cingulate cortex to form a scaffold for the callosal axons 5 that approach from cortical layers II, III, and V. 11 Both cingulate and callosal axons express NRP1 receptors, thus, Sema3C interacts with both. Because Sema3C is highly concentrated at the midline, it must be regulated to ensure proper axon crossing to the opposite cortex. After callosal axons cross the midline, EphB1 is upregulated and colocalizes with NRP1 to silence Sema3C attraction postcrossing. 7
Another guidance cue/receptor, Netrin-1/DCC, attracts CC axons to the midline
A second guidance cue, Netrin-1, is also expressed at the cortical midline and again both callosal and cingulate axons express its receptor, deleted in colorectal cancer (DCC).12,13 In addition to DCC, callosal axons express Robo receptors. The Netrin-1/DCC pair functions in CC development by directly attracting cingulate axons and indirectly modulating Slit/Robo repulsion of callosal axons.13,14 Again, we see a change in gene expression that regulates pathfinding. DCC is highly expressed before axons cross the midline and is downregulated postcrossing, allowing Robo1 to take over guidance. 13
Repulsive Slit/Robo ensures target acquisition
The Slit/Robo guidance cue/receptor pair repels callosal axons. Slit2 is secreted by two midline glial populations: the glial wedge, which lies inferior to the CC, and the indusium griseum, which is present superior to the developing CC. Once callosal axons have been attracted to the midline by Sema3C/NRP1 signaling, Slit/Robo repels the axons into the opposite cortex where they continue to their proper target. 12 When functioning properly, Slit/Robo repulsion prevents ectopic axon growth into the septum.6,12
CC agenesis seen with lack of GABAergic guidepost cells
Without the CC properly formed, particularly in cases of agenesis where the CC is entirely absent, development is delayed in motor, speech, and social function. 15 In addition to these neurological deficits, damage to the CC can also result in aberrant behavior. 15 Abnormalities associated with guidance mechanisms significantly impact CC development. Niquille et al. investigated how the CC develops without GABAergic guidepost neurons present. To do this, they inhibited Mash1, a transcription factor expressed in GABAergic progenitors that produce cortical interneurons and observed partial to severe agenesis of the CC. Guidance cues (L1CAM, NRP1, DCC) were expressed at normal levels but axons were misguided, evidenced by Probst bundles along the midline. 5 The Probst bundle phenotype is characterized by aberrant callosal axon growth into the septum or along the midline. The fact that callosal axons were misguided indicates that GABAergic neurons provide a necessary structural mechanism for guidance. As evidenced by these mutants, guidepost cells physically direct callosal axons to form the CC in addition to release guidance cues.
Sema3C/NRP1 signaling mutations cause misguided cingulate and callosal axons
Sema3C guidance cue and Nfib transcription factor mutations that disrupt signaling through NRP1 receptors cause CC agenesis. In a Sema3C null mutant study, instead of crossing, callosal axons formed Probst bundles along the midline.5,12 Sema3C null mutants showed partial agenesis where no callosal axons crossed dorsally, and only some crossed ventrally. 5 This study suggests that NRP1 is important for dorsoventral organization, specifically, guiding dorsal CC axon crossing. 5 Furthermore, in mutants where NRP1 is unable to bind semaphorins, cingulate axons were also misguided and formed ectopic Probst bundles. 4 The Nfib transcription factor is widely expressed in the cingulate cortex where NRP1-expressing pioneer axons originate. 8 In Nfib− deficient mice, NRP1 receptor expression was diminished and cingulate axons did not cross the midline, causing callosal agenesis. 8
Netrin-1 and DCC mutants both display CC agenesis
Both Netrin-1 and DCC mutants also lack a CC due to effects on guidepost cells. 6 Septal fusion is a prerequisite for CC formation, 16 and guidepost populations are vital in this region to facilitate axon crossing. In Netrin-1 mutants, callosal guidepost populations have defects and septal fusion does not occur, contributing to agenesis. 16 Together, these two abnormalities in Netrin-1 mutants highlight signaling processes that are possibly disrupted during agenesis. 16 In addition, DCC mutants do not respond properly to draxin, an axon guidance protein that binds these receptors, and they exhibit uninhibited axonal outgrowth and reduced growth cone collapse. 17 This could cause callosal axons to improperly grow into the septum or other locations.
Distinct CC defects are exhibited in Slit/Robo mutants
Robo2 mutants exhibit partial to severe CC agenesis; 12 both Robo1 and Robo2 mutants display Probst bundles along the midline.12,18 In addition, in Robo1 mutants, callosal axons improperly mix with hippocampal commissural axons at the midline, indicating a potential role for Robo1 in ensuring that these axon populations remain discrete. 18 The nuclear family 1A-type (Nfia) transcription factor also regulates CC formation. 19 Nfia mutants display absence or reduction of GFAP+ glia in the indusium griseum and glial wedge and reduced Slit2 expression in these populations. 20 Without Nfia, agenesis occurs, emphasizing a role for this transcription factor both in midline glial cell development and guidance cue secretion. 20
Aberrant guidepost cells could underlie CC defects in FASD
Severe cases of FASD consistently present with CC agenesis and other CC abnormalities. These defects are recapitulated in animal studies, such as the improper formation of the splenium CC area seen in rats prenatally exposed to alcohol. 21 These abnormalities mimic phenotypes displayed in mutants of CC axon guidance mechanisms, suggesting that alcohol may act by disrupting guidance molecules and guidepost cell populations.
Alcohol is well known to affect the delicate balance between synaptic excitatory and inhibitory signals, maintained in part by GABAergic and glutamatergic neurons by decreasing inhibition and increasing excitation. 22 Several studies show that alcohol reduces the number of GABAergic neurons in the cerebral cortex, anterior cingulate cortex, hippocampus, and amygdala.23,24 Contrary to these findings, other studies using a low dose exposure paradigm to induce FASD found increased neurogenesis in the MGE, where GABAergic neurons originate, which stimulated more interneurons to migrate to the medial prefrontal cortex.25,26 Since these neuronal populations play a key role in physically and chemically guiding callosal axons across the midline as guidepost cells, any abnormalities in these cells would impact CC formation. As a well-established target of alcohol exposure, GABAergic neuron pathology could explain why we see CC agenesis and Probst bundles in FASD. More GABAergic cells at the midline would block pioneer and callosal axons from crossing, forcing growth along the midline as Probst bundles. Fewer GABAergic cells could cause improper cortical innervation. Studies in macaque monkeys show this potential result, as alcohol exposure increased the number of axons in the CC with the rostral portions being affected most significantly. 27 We are not aware of any studies investigating the effects of alcohol on glial populations that serve as guidepost cells, but glial cells are a well-established target of alcohol pathology and have been hypothesized to underlie CC dys/agenesis. 28
Alcohol exposure damages the cingulate cortex, the origin of pioneer axons for the CC
Several studies show that alcohol directly affects the cingulate cortex. A rat study, with a range of alcohol exposure doses, found resulting microhemorrhages in the brain that led to neuronal loss concentrated in the cortex, including the cingulate cortex. 29 Supporting this finding, structural MRIs show that alcohol-exposed individuals had reduced overall volume, grey matter, and white matter in the cingulate gyrus, one component of the cingulate cortex, as well as the CC. Despite controlling for overall change in volume, white matter presented significantly reduced compared with controls. 30 Another study focused on the anterior cingulate cortex also found reduced surface area in alcohol-exposed individuals. 31 Decreased cingulate volume and damage to the area can negatively impact the number and function of pioneer axons that form a scaffold for callosal neurons to cross the midline. Without enough pioneer axons, callosal axons would struggle to find and follow the proper path, further affecting the formation of the CC.
Alcohol alters expression of semaphorin guidance receptors, EphB1 and NRP1 that regulate CC formation
In relation to guidance cues, postnatal alcohol exposure downregulates EphB1 receptors in mice. 32 If this is also the case prenatally, alcohol could cause aberrant callosal axon growth as Probst bundles given that Sema3C attraction would not be appropriately silenced (Figure 1). Accordingly, Probst bundles are seen alongside severe FASD with complete or partial CC agenesis. 15 Tsai et al. investigated microRNA miR-153 and its target transcripts which include Nfib in neural stem cells and found that miR-153 suppresses Nfib. 33 Alcohol exposure broadens the range of Nfib expression, likely due to repression of miR-153 expression. 33 Alcohol-induced Nfib expression could upregulate NRP1, as their expression may be linked. 8 NRP1 upregulation would make cingulate and callosal neurons hypersensitive to Sema3C. Such perturbations in Sema3C pathway signaling and expression could contribute to CC abnormalities observed in cases of FASD.
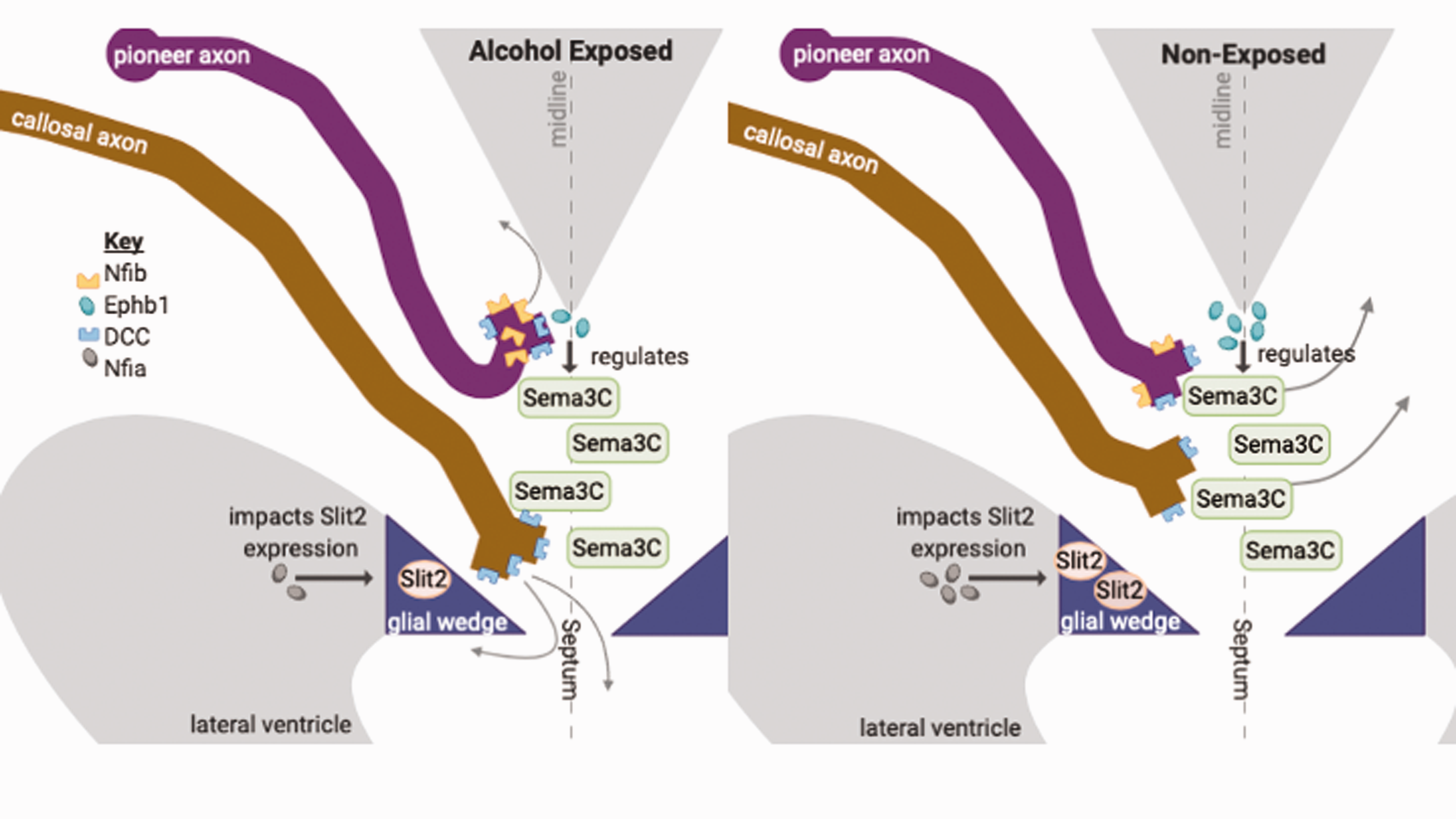
Effects of alcohol on axon guidance-associated mechanisms in the Corpus Callosum (CC). This schematic depicts underlying mechanisms for corpus callosum agenesis and Probst bundles displayed in Fetal Alcohol Spectrum Disorders (FASD). Semaphorin (Sema) 3C attracts axons toward the midline and Slit2 expressed by the glial wedge repels axons to the opposite cortex. Alcohol downregulates Nfia (grey) and EphB1 (teal) expression and upregulates Nfib (yellow) and DCC (blue) expression. These effects can lead to abnormal corpus callosum development often seen in FASD. Schematic adapted from literature.6,11 (A color version of this figure is available in the online journal.)
Effects of alcohol on Netrin-1/DCC Signaling may mediate CC defects
Several studies indicate that alcohol disrupts Netrin-1/DCC signaling and function34,35 which may contribute to CC agenesis or other abnormalities commonly observed with FASD. Alcohol, even at low concentrations, inhibited Netrin-mediated growth cone collapse, suggesting that alcohol may render growth cones unresponsive to its repulsion. 34 Another study found that both binge and chronic alcohol exposure upregulated DCC 35 which could override Slit/Robo signaling and prohibit callosal axon crossing to the opposite cortex causing midline Probst bundles (Figure 1). Though these studies looked at Netrin-mediated repulsion, it is also plausible that alcohol disrupts Netrin-mediated attraction which would interfere with the ability of pioneering cingulate axons to form a scaffold for callosal axons.
Alcohol downregulates transcription factor Nfia, reducing Slit2 gene expression
Alcohol exposure downregulates Nfia, 19 which in turn downregulates Slit2 expression, garnering Slit2 unable to repel axons properly. These effects would make it more difficult for callosal axons to reach their proper targets in the opposite cortex (Figure 1). We believe that advances in treatment for FASD would follow by taking a closer look at the overlapping mechanisms contributing to CC abnormalities observed in mutant and alcohol studies. Because the Slit/Robo pathway is actively being pursued as a therapeutic target in cancer research, it is likely that pharmaceuticals will soon be available36,37 which could serve dual purpose as FASD treatments.
Cerebellum—Mossy and Climbing fibers
Semaphorin signaling drives cerebellar functional connections
The cerebellum consists of three major layers: the granule cell layer containing mossy fibers which synapse on granule cells, the Purkinje cell layer containing climbing fibers which synapse on Purkinje cells, and the molecular layer containing basket cells which regulate Purkinje cell communication with deep cerebellar nuclei. 38 During development, Purkinje cells are guided to the deep cerebellar nuclei and then to the cortex to relay sensory signals for movement and balance.39,40 Climbing fibers originate in the olivary nucleus of the brainstem and mossy fibers originate in the hindbrain and spinal cord where they will then travel to the cerebellar rhombic lip to migrate through the three layers. 41 Figure 2 illustrates the organization of cerebellar circuitry. Disruption and under-development of the cerebellar circuitry cause motor deficits including altered balance and motor coordination. 40 The guidance cue/receptor pair Sema3A/NRP1 repels climbing and mossy fibers away from the cerebellar midline, aka vermis, to their targets. 42 Plexin-A3, another semaphorin receptor, also repels mossy fibers. 43 It signals with Sema3C, but these interactions are not well studied. Jiang et al. recently found Plexin-A3 conjugates with collapsin response mediator protein 2 (CRMP2) to promote dendrite growth of cerebellar granule cells. 43 CRMP family members promote cytoskeleton assembly via their interactions with actin and tubulin which may impact growth cone collapse caused by semaphorin signaling. 43

Effects of alcohol on organization of cerebellar circuits. Climbing and mossy fibers enter the cerebellum and synapse on Purkinje and granule cells, respectively. Illustrated are the three layers of the cerebellum: the granule cell layer, Purkinje cell layer, and the molecular layer. The Purkinje axons then synapse on the deep cerebellar nuclei. Guidance cue/receptor families that guide these axons are concentrated at the midline, namely Slit/Robo, Netrin-1/DCC, and Semaphorin (Sema) 3A/Neuropilin (NRP)1. Due to the effects of alcohol on these signaling pathways, there could be a disruption in the direction and organization of mossy and climbing fibers synapsing on the granule and Purkinje cells. A: anterior; P: posterior; M: medial; L: lateral. Schematic adapted from literature. 83 (A color version of this figure is available in the online journal.)
Netrin/DCC and Slit/Robo signaling cooperate to guide and organize cerebellar axons at the midline
Climbing and mossy fibers express DCC and Robo2 receptors and are thus both affected by Netrin-1 and Slit signaling. 44 Netrin-1 and Slit ligands are concentrated in the midline; however, Netrin-1 attracts climbing and mossy fibers, while Slit antagonizes this attraction by repelling climbing and mossy fibers away from the midline.44,45 How these two pathways cooperate at the midline to regulate cerebellar axon guidance is poorly understood.
Slit may cooperate with semaphorins and neural cell adhesion molecules in cerebellar guidance
Recent work by Dominici et al. utilized a conditional knockout strategy to remove Robo1 and Robo2 from precerebellar neurons whose axons make up mossy and climbing fibers. 46 Their work concluded a non-cell autonomous function for midline Slit repulsion. 46 They propose that instead of working through Robo receptors, Slit may cooperate with plexinA1, a semaphorin receptor, or neural cell adhesion molecules (NrCAMs). 46
Mutations in Sema3A signaling alters basket cell axon pathfinding, disrupting inhibitory cerebellar output
Basket cells project onto Purkinje cells forming the inhibitory pinceau synapse at the Purkinje cell layer 47 (Figure 2). A cell adhesion molecule in the cerebellum, neurofascin186 (NF186) is expressed on both cell types, enabling pinceau synaptic transmission between them. 47 It is hypothesized that Sema3A stabilizes expression of the NRP1 receptor to trigger interaction with NF186 on the surface of cerebellar axons which aids in their pathfinding. 47 Sema3A and NF186 mutations alter axon guidance and connectivity during cerebellar development. 47 Sema3A mutants display impaired pinceau synapse formation and disrupted basket cell organization in the Purkinje cell layer. 47 In another study, Cioni et al. showed that either lack of Sema3A or inhibiting Sema3A binding domains on NRP1 receptors reduced basket cell axon branching in the Purkinje cell layer. 42 Removing NF186 also disrupts basket cell organization in the molecular layer, altering axon guidance. 47 The study concluded that Sema3A/NF186 interaction is required for proper basket cell guidance and target recognition. 47
Loss of Netrin-1 or its receptor uncoordinated-5 (UNC5) cause disorganization in the cerebellum
Complete knock-out of Netrin-1 is lethal and causes severe axon defects in the spinal cord. 48 The spinal cord is one origin of mossy fibers and secretion of Netrin from the dorsal spinal cord is required to guide mossy fibers to the cerebellum. 48 Thus, Netrin deficiency in the spinal cord could impact mossy fiber organization in the cerebellum. At the cerebellar midline, UNC5 receptors bind with Netrin-1 and repel cerebellar axons away from the midline. 45 Mutants of the UNC5H3 isoform displayed decreased cerebellar midline volume, 49 specifically, Purkinje and granule cell arrangements in their respective layers of the rostral cerebellum were abnormal. 49 Furthermore, random scattering of granule cells was observed among the layers. 49 Disorganization of granule cells among the three layers of the cerebellum would disrupt the specialized functions of each layer along with the guidance cues that direct each axon to its synaptic target. 49
Slit/Robo prevents Purkinje dendritic self-crossing which is important for proper motor function
In the cerebellum, the repulsive force of the Slit2/Robo2 guidance cue/receptor pair aids in self-avoidance of cerebellar dendrites in the Purkinje layer so that proper climbing fiber synaptic connections can be made. 50 Gibson et al. observed that the deletion of either Slit2 or Robo2 in the cerebellum causes dendrites to self-cross. 50 When Robo2 was specifically deleted from Purkinje cells, they also observed gait alterations, likely due to redundant synaptic connections. 50
Alcohol inhibits L1CAM-mediated axonal outgrowth in the cerebellum
A NrCAM affected by alcohol, L1 cell adhesion molecule (L1CAM), plays a role in axon migration, neurite extension, and neuronal guidance in the cerebellum and other key brain regions. 51 Treatment of cultured cerebellar granule cells with alcohol inhibited L1CAM-mediated axonal outgrowth and guidance. 51 This inhibition could result from lower expression, lower cell surface distribution, and/or impaired interaction with the cytoskeleton. 51 Bearer’s lab discovered that alcohol disrupts L1CAM phosphorylation/dephosphorylation and increases its association with lipid rafts in vivo. 51 The phosphorylation status of L1CAM is critical for L1-mediated neurite outgrowth. 52 Since granule cell migration requires Semaphorin interaction with L1CAM, 53 it would be interesting to explore whether Semaphorin signaling has any impact on alcohol-induced inhibition of L1CAM-mediated axonal growth and guidance in the cerebellum. Alcohol-induced alterations in Semaphorin signaling could lead to disorganized mossy fiber distribution directly or indirectly via disorganized granule cell migration. Alcohol targeting of L1CAM or its interacting partners could explain abnormalities in brain morphology that are evident at the cerebellar midline in severe cases of FASD. 52 Developmental alcohol exposure increases basket cells and their innervation of Purkinje cells, altering cerebellar output 54 which could also result from effects on Semaphorin signaling. Further study of the effects of alcohol on molecular players driving these connections is warranted.
Alcohol targets cytoskeletal genes that are critical for axon growth and guidance
Alcohol targets the cytoskeleton, which is key in several developmental processes including growth cone responses.55,56 Cytoskeletal components actin and tubulin are involved in growth cone responses during pathfinding. 57 In the cerebellum, downstream of Netrin-1 signaling, the tubulin beta 3 class III (TUBB3) gene plays a key role in axon branching and outgrowth. 57 Recently, the TUBB3 gene was identified as an alcohol target. 58 TUBB3 is essential for Netrin-1 repulsion. 57 Thus, since TUBB3 is susceptible to alcohol exposure, cytoskeletal dynamics could be a converging mechanism for the effects of alcohol on axon guidance signaling.
Motor deficiencies observed with FASD can be attributed to altered Slit/Robo pathways
Given the motor deficiencies observed with FASD, we hypothesize that alcohol exposure mimics the self-crossing defects evident with reduced Slit/Robo guidance that affects the integrity of the Purkinje dendritic arbor and climbing fiber function. Studies investigating these alternate mechanisms for Slit signaling in the cerebellum could further illuminate how their disruption possibly contributes to phenotypes observed with FASD.
Hippocampus—Mossy fibers
Part of the limbic system, the hippocampus is involved in processing learning, memory, and emotions. The hippocampal formation consists of the dentate gyrus (DG), the Cornu ammonis areas (CA), the presubiculum, the parasubiculum, and finally its connection to the entorhinal cortex (Figure 3). Granule cell axons, also known as mossy fibers, originate in the granule cell layer of the DG and synapse with CA3 pyramidal neurons. 59 In addition to short-term memory processing, mossy fibers are associated with seizure susceptibility and neurodegeneration. 59

Effects of alcohol on mossy fiber projections within the hippocampus. Granule cell axons, or mossy fibers, arise from the DG and are guided towards CA3 by Netrin-1 attraction and Sema3A repulsion to synapse with CA3 pyramidal neurons. Netrin-1 in the CA3 attracts mossy fibers while Sema3F in the CA1 repulses them ensuring proper guidance of the mossy fibers. When exposed to alcohol these guidance mechanisms are negatively impacted leading to the aberration of mossy fibers. Adapted from literature. 84 (A color version of this figure is available in the online journal.)
Semaphorin signaling guides mossy fiber circuit development
Semaphorin signaling of mossy fibers in the hippocampus is two-fold: Sema3A repels mossy fibers towards the CA3 and prevents them from moving back into the DG 59 while Sema3F uniquely signals with Plexin-3A and neuropilin-2 (NRP2) receptors to repel mossy fibers from the CA1 region towards the CA3 region. 59 Additionally, Sema3F/NRP2 are involved in pruning mossy fiber synapses on CA3 axons. 60 When granule cell axons synapse onto their own dendrites, mossy fiber sprouting occurs, creating excitatory autosynapses, instead of targeting the CA3. 61
Netrin-mediated repulsion also regulates mossy fiber sprouting
As in the cerebellum, the activity of Netrin-1 in the hippocampus as attractive or repulsive depends on the receptor it is signaling through: DCC mediates attraction and UNC5 repels. 62 When both receptors are expressed, UNC5 appears to be dominant to DCC resulting in repulsion. 63 Hyperexcitability causes mossy fiber sprouting due to upregulation of Netrin-1/UNC5-mediated repulsion. 62
Slit and Robo may guide hippocampal connections and regulate synaptic plasticity
There is little known about Slit and Robo expression patterns and their guidance in the hippocampus is not well researched. Slit1 and Robo1 mRNAs expression is greatest in the granule cell layer of the DG and in CA2-CA4 regions. 64 Slit2 and Robo2 are also uniformly expressed through all CA regions. 64 Slit2 repels hippocampal axons from DG explants and collapses their growth cones, presumably due to Robo1 expression in the DG. 65 Although the DG completes development postnatally these studies provide evidence that Slit2 may possibly follow a similar pattern of repulsing mossy fiber afferents in order to segregate them from crossing over entorhinal cortex and the telencephalic midline. 65 It has been recently suggested that, in addition to traditional roles of guidance, Slit and Robo may be important for hippocampal synaptogenesis and synaptic plasticity in vivo. 64 These roles in cognitive function are thus potentially vulnerable to the effects of alcohol exposure.
Mossy fiber guidance by Semaphorin signaling is important for hippocampal function
Research shows that irrespective of whether Sema3F is removed from all cells, or pyramidal neurons or granule cells specifically, its loss causes abnormal mossy fiber growth and guidance. 66 Likewise, these defects are mimicked in NRP2 receptor mutants.66,67 Recent research on semaphorin signaling in the hippocampus focuses on the behavioral consequences of impaired Sema3F and explores the role of Sema3A in mossy fiber pathologies. For example, Sema3F mutant mice show decreased locomotion, enhanced fear, and increased anxiety suggesting that Sema3F signaling plays a role in the development of these circuits. 68 Sema3A down regulation causes mossy fiber sprouting. 61 This phenomenon is linked to increased excitability in epilepsy; 69 however, whether it is a result of or causes epilepsy is unclear. 70
Prenatal alcohol exposure negatively impacts mossy fiber organization and hippocampus synaptogenesis
Despite minimal studies, there are direct effects of alcohol on mossy fiber development and neuronal circuits. Studies of cultured rat hippocampal pyramidal cells have shown that alcohol inhibits dendrite development by decreasing their length and quantity within a cell and producing a secondary effect of inhibiting synaptogenesis. 71 Likewise, in rat prenatal alcohol exposure studies, an abnormal distribution of mossy fiber axons was found in the temporal region concluding that alcohol also disturbs the organizational processes of mossy fiber axons. 72
Effects of alcohol on Semaphorin signaling in the hippocampus may contribute to characteristic FASD features
Behavioral phenotypes such as decreased locomotion, increased fear and anxiety, and hyperexcitability that consistently present in individuals with FASD 73 also result from lack of semaphorin isoforms, specifically Sema 3A and Sema 3F. 68 As in the cerebellum, Semaphorins interact with NrCAMs to guide hippocampal axons. Sema3A interactions with L1CAM underlie the ability of septal axons to target the hippocampus and this guidance is disrupted by alcohol exposure. 74
In hippocampal neurons, CRMP2 interacts with microtubules to specify axons and promote axon growth in hippocampal neurons. 75 Alcohol exposure can increase CRMP2 levels in the hippocampus, 76 which we expect would result in supernumerary axons. 75 In the cerebellum, CRMP2 is downstream of Sema3C/PlexinA3 signaling. 43 Given the influence of hippocampal mossy fibers on seizure susceptibility, coupled with the observation that children with FASD present a high prevalence of epilepsy, 77 we propose that effects of alcohol on Semaphorin signaling could be a causal link resulting in guidance errors that disrupt circuitry in this region. In support of this hypothesis, lacosamide, an antiepileptic drug, prohibits CRMP2-mediated microtubule assembly which underlies addiction-like behavior. 76 Given mounting evidence that FASD is associated with increased propensity for substance use disorders, 78 this could be a critical mechanism to investigate.
Alcohol-induced changes in gene expression during early brain development could impact axon guidance
Alcohol consumption during pregnancy changes gene expression in the developing fetal brain.32,58 Recent work shows that the genes encoding each guidance cue/receptor family discussed herein and several of their interacting partners are differentially affected by alcohol exposure in the rostroventral neural tube that develops into midline structures. 58 Though most of these genes are only affected transiently, 58 changes in their expression reveal the widespread, adverse effects of alcohol exposure in early development. We stress the importance of studying the vulnerability of these genes to alcohol toxicity to advance FASD treatment.
Conclusions
The functional consequences of FASD reduce the quality of life for affected individuals due to lifelong physical, mental, behavioral, and learning disabilities. Emerging evidence suggests breeches in white matter integrity are a hallmark of FASD, irrespective of facial dysmorphology. 79 Of the brain regions discussed herein, most of our knowledge pertains to mechanisms underlying CC formation, but there are several other known effects of alcohol that can induce agenesis. These effects include apoptosis during gastrulation that influences early stages of brain development that form these cortical brain regions.80,81 Alcohol-induced apoptosis throughout the brain, particularly in cortical neurons, also suppresses neuronal activity and provides another mechanism to explain agenesis in FASD. 82 There are also many studies investigating the effects of alcohol on glial cells, but none in relation to CC formation. Nevertheless, as in other brain regions, the effects of alcohol in concert with known mechanisms are under-explored. It is noteworthy that L1CAM mediates guidance for each of the neural pathways we have discussed and is affected by alcohol, so may be a productive starting point. We posit that alcohol targets axon guidance mechanisms as a unifying explanation for the multifaceted abnormalities we see in all stages of development that characterize FASD. It is imperative to study the effects of alcohol on these mechanisms to demonstrate a causal link between developmental alcohol exposure and impaired connectivity, with the ultimate goal of discovering treatments for the cognitive and behavioral deficits in individuals with FASD.
Because the studies referenced here were done in cell culture, we believe that future studies should prioritize in vivo investigations of the effects of alcohol on the timing and expression of guidance cue/receptor families. Although single cell cultures cannot accurately recapitulate the dynamic environment of the developing brain, they do allow dissection of mechanism which is critical to understand in vivo observations. Given our current understanding, it is highly defensible that alcohol-induced changes in patterning, expression, and timing of axon guidance signaling mechanisms contribute to the altered neural connectivity observed in FASD. Further study is needed to elucidate and understand the complex influences that alcohol has on the developing brain which will thereby lead to the development of more effective treatments for FASD.
Footnotes
ACKNOWLEDGEMENTS
We are extremely grateful to Dr Quinn Smith for her careful read of the manuscript as well as her genuine curiosity and support of our work.
AUTHORS’ CONTRIBUTIONS
All authors participated in researching, drafting, and revising the manuscript.
DECLARATION OF CONFLICTING INTERESTS
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
FUNDING
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by NIAAA grant #1R21AA025740-01A1 to C.F.