
Abstract
Age-related macular degeneration is the leading cause of blindness in the elderly. The Y402H polymorphism in complement factor H promotes disease-like pathogenesis, and a Cfh+/− murine model can replicate this phenotype, but only after two years. We reasoned that by combining CFH deficiency with cigarette smoke exposure, we might be able to accelerate disease progression to facilitate preclinical research in this disease. Wild-type and Cfh+/− mice were exposed to nose-only cigarette smoke for three months. Retinal tissue morphology and visual function were evaluated by optical coherence tomography, fundus photography and autofluorescence, and electroretinogram. Retinal pigment epithelial cell phenotype and ultrastructure were evaluated by immunofluorescence staining and transmission electron microscopy. Cfh+/− smoking mice showed a dome-like protruding lesion at the ellipsoid zone (drusen-like deposition), many retinal hyper-autofluorescence spots, and a marked decrease in A- and B-wave amplitudes. Compared with non-smoking mice, wild-type and Cfh+/− smoking mice showed sub-retinal pigment epithelium complement protein 3 deposition, activation of microglia, metabolic waste accumulation, and impairment of tight junctions. Microglia cells migrated into the photoreceptor outer segment layer in Cfh+/− smoking mice showed increased activation. Our results suggest that exposing Cfh+/− mice to smoking leads to earlier onset of age-related macular degeneration than in other animal models, which may facilitate preclinical research into the pathophysiology and treatment of this disease.
Impact statement
Age-related macular degeneration (AMD) is by far the leading cause of irreversible visual impairment in people over 60 years old, and there is no effective therapy for dry AMD. A mature animal model is of great significance for the study of this disease. Our study has generated a novel AMD murine model that combines known genetic and environmental risk factors to produce a relevant tool to analyze the pathophysiology and potential treatment of the disease in its early stages.
Introduction
Age-related macular degeneration (AMD) is by far the leading cause of irreversible visual impairment in people over 60 years old. In early stages of the disease, drusen and retinal pigment epithelium (RPE) dysfunction are observed, whereas the advanced stage involves either geographic atrophy or neovascularization. Most patients (80%) have non-exudative or dry AMD, for which there is no effective therapy. 1 The pathogenesis of AMD is complex, and it involves genetic polymorphisms and environmental factors. Currently, more than 50 susceptibility genes have been identified, among which CFH and ARMS2 genes are the most important in pathogenesis. 2 Environmental risk factors include smoking, unhealthy diet, obesity, high blood pressure, hypercholesterolemia, and antioxidant deficiency. The Y402H polymorphism is the single nucleotide polymorphism of CFH. This mutation may impair the binding efficiency and other molecule function, and hence reduce the inhibitory abilities of CFH, resulting in the over-activation of complement alternative pathway at the interface between RPE and Bruch’s membrane. Epidemiological studies suggest that carriers of the CFH Y402H mutation and heavy smokers are at higher risk of AMD. 3 Multiple studies have confirmed the association between Y402H polymorphism in CFH and increased risk of AMD.4,5
Although mice do not have a macula, their retinal structures are similar to those of the peripheral retinas in humans, and they can exhibit the characteristics of dry AMD. 6 Deleting or decreasing expression of the Cfh gene predisposes mice to AMD.7–10 Cfh–/– mice show significantly greater stress responses and inflammation in an open environment than in a pathogen-free environment. 11 These findings suggest that when CFH is deficient, environmental risk factors may contribute to a more severe phenotype.
Cigarette smoke, which increases the risk of AMD by two to four folds, 12 is the leading preventable risk factor, and it synergizes with mutations in certain genes to further increase risk of the disease. 13 Several groups have described a mouse model in which AMD can be initiated through cigarette smoke exposure,14–16 an effect that involves activation of the complement system. 17 Indeed, complement inhibitors can prevent and even repair smoking-induced ocular injury. 18 These findings suggest that there is a close association between cigarette smoke exposure and complement activation.
Studies with the previously described mouse models of AMD can take a long time, since two years may be needed for the disease phenotype to manifest. 8 We reasoned that combining excessive complement activation with cigarette smoke exposure might accelerate development of the disease in mice. If so, the resulting animal model could support more efficient and extensive preclinical studies into disease onset, progression, and treatment.
Materials and methods
Generation of Cfh+/ – mouse
The CRISPR/Cas-9 system was used to generate homozygous Cfh knockout (Cfh–/–) mice. Two single-guide RNAs were designed to target exons 2 and 3 in the mouse Cfh gene (ENSMUSG00000026365): 19 sgRNA1, CCGTATATGAACGGATGATCAGG; and sgRNA2, TCATCCTACGTAGGATTAGCAGG. The sgRNAs were synthesized and inserted into the Cas9 vector plasmid (pRP [CRISPR]-Hcas9-U6, VectorBuilder, China), then transformed into E. coli and amplified. The amplicons were purified for DNA sequencing. The linearized DNA was transcribed and amplified into RNA, and the purified RNA was identified by electrophoresis. The diluted RNA was microinjected into mouse fertilized eggs, and then the eggs were transferred into pseudopregnant female C57BL/6 mice. After giving birth to mice, F1 generation mice were obtained. Tail tissue was harvested for Sanger sequencing and PCR genotyping. The primers used were F, ATGTGTTTAAGCCCAAATCTGCTCC; Cfh-R, GAGGCAACAATGAGTTCAAGAAACCA; and He/Wt-R, AACTTCTTCTTCTCCCTCGCCCAT. All wild-type (WT) C57BL/6 mice were purchased from Beijing HFK BioScience (Beijing, China).
Mice were housed on a 12-h light/dark cycle and received food and water ad libitum. All procedures were conducted in compliance with the Association for Research in Vision and Ophthalmology (ARVO) statement and approved by the Animal Ethics Committee of West China Hospital of Sichuan University (approval number: 2019280 A).
Grouping of animals and treatments
Since the complement system of Cfh–/– mice may be deficient, 9 the mice can develop severe membranoproliferative glomerulonephritis, leading to death at 12 to 24 months. 19 Therefore, Cfh+/− mice were selected for this study. Estrogen level affects the formation of subretinal deposits, 20 so all mice in this study were male. Eight-week-old male mice were randomly divided into smoking or non-smoking groups for a total of four conditions: WT non-smoking (n = 10), WT smoking (n = 20), Cfh+/− non-smoking (n = 10), and Cfh+/− smoking (n = 20). Smoking groups were exposed to smoke using a nose-only cigarette smoke exposure method as described. 21 Briefly, the mouse was restrained in a custom-designed container, and the container was mounted in a smoking chamber in which the cigarette smoke flowed in one direction. There were closed-pipe connections linking the smoking chamber, the fixed-rate pump (CH Technologies, Westwood, NJ, USA), and the Baumgartner-Jaeger CSM2082i automated cigarette smoke machine (CH Technologies, West-Wood, NJ, USA). Commercially available cigarettes (Marlboro, Philip Morris, USA) were used to generate smoke. The cigarette smoke was diluted with fresh air (2.6:9.1) to obtain a concentration of total suspended particulates of 250 mg/m3. The mice were exposed to cigarette smoke two times/day for 75 min/time during 12 weeks. Non-smoking mice were maintained under standard husbandry conditions for 12 weeks.
OCT, fundus photography, FAF examination, and ERG
Mice were sedated with an intraperitoneal injection of chloral hydrate (4 μl/g). Then, tropicamide eye drops (Santen Pharmaceutical, Japan) and oxybuprocaine hydrochloride eye drops (Santen Pharmaceutical) were administered to provide pupil dilatation and topical anesthesia, respectively. OCT, fundus photography, and FAF examinations were performed using the Spectralis imaging platform (Spectralis HRA + OCT, Heidelberg, Germany). Images were analyzed for fluorescing spots using ImageJ (http://imagej.net/ImageJ, version 1.52a, USA); the number of fluorescing spots of one retina was manually counted. Retinal function was evaluated using flash ERG (RetiMINER IV, IRC, China). Mice were dark-adapted overnight before anesthesia and pupil dilatation, and experiments were performed under dim red illumination.
Immunofluorescence staining
Mice were sacrificed by cervical dislocation under general anesthesia. Eyes were enucleated immediately, then a hole was punched at the limbus, and the punch biopsy was fixed in 4% paraformaldehyde for 40 min at room temperature. After dehydration and embedding at optimal cooling temperature, cryo-sections were made at a thickness of 6 µm. Immunofluorescence staining was performed for C3b/iC3b/C3c, IBA1, rhodopsin, and IB4. The sources and dilutions of all antibodies are listed in Table S1. Images were captured using the Axio Imager Z2 fluorescence microscope (Zeiss, Germany). RPE-choroid-sclera flat-mounts were prepared 22 for ZO-1 immunofluorescence staining, and an A1RMP + confocal microscope (Nikon, Japan) was used to capture images. For quantitative analysis, the number of activated microglia cells was manually counted using ImageJ software.
Transmission electron microscopy
Mice were sacrificed, eyes were enucleated, and a hole was punched at the limbus using a 29 G needle. Eyeballs were fixed in electron microscopy fixative (Servicebio, China) at 4°C. The fixed tissues were cut into 1-mm3 cubes on clean filter paper. After osmium fixation, acetone gradient dehydration, embedding agent infiltration, and polymerization, the tissue pieces were cut into 70-nm sections, which were then double-stained with uranium acetate and citric acid. Images were obtained using an H7650 TEM (Hitachi, Japan).
Statistical methods
Numerical data were processed using Microsoft Excel (version 2013), and graphs were generated using GraphPad Prism 6.0 for Windows (GraphPad Software, Bethesda, MD, USA). Appropriate statistical methods were selected according to the type of data. For the comparison of the number of fluorescing spots between Cfh+/− smoking and Cfh+/− non-smoking mice, an independent-samples t-test with unequal variances was chosen. ANOVA with post hoc Dunnett T3 or Tukey HSD test was chosen to assess differences in the numbers of activated microglia cells and the ERG amplitudes among the four groups. Statistical analyses were performed using SPSS 13.0 for Windows (SPSS, Chicago, IL, USA), and differences associated with P < 0.05 were considered significant.
Results
Genotyping of Cfh knockout mice
Cfh knockout was confirmed by Sanger sequencing in mice of the first filial (F1) generation, and we determined the genotype of the resulting offspring by polymerase chain reaction (PCR) and agarose gel electrophoresis (Figure 1). The results indicated that the CRISPR-Cas9 system was effective in knocking out the Cfh gene. Cfh+/− mice were used in all of the following experiments.
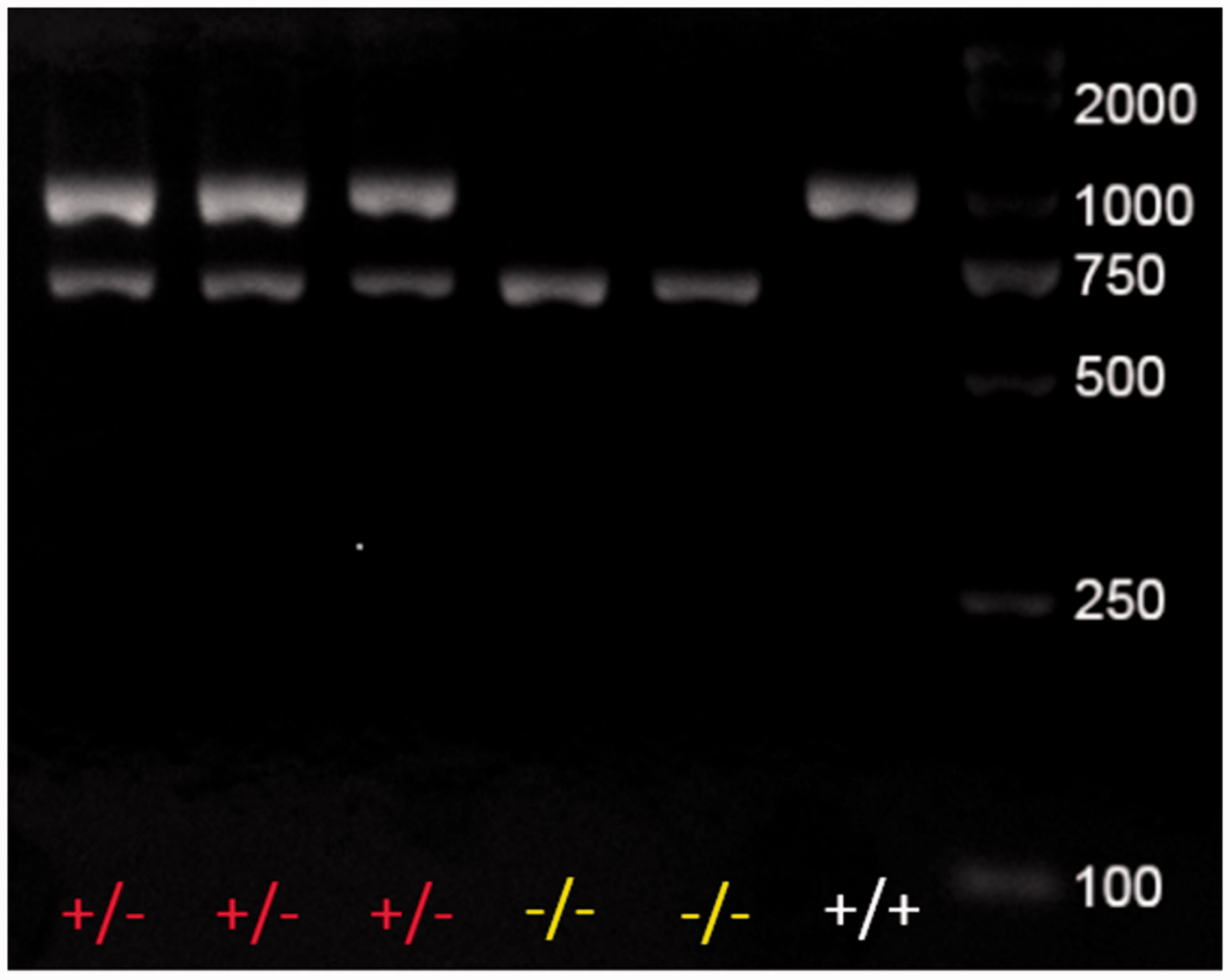
Genotyping of mice. Polymerase chain reaction (PCR) and agarose gel electrophoresis to genotype the progeny. (A color version of this figure is available in the online journal.)
Smoking and CFH deficiency impaired visual function
Photoreceptor degeneration and retinal function were assessed using electroretinography (ERG), which is a useful tool in the early diagnosis of AMD.23,24 Decreased A- and B-wave amplitudes are one of the hallmarks of early AMD. 25 The ERG responses of all mouse groups under scotopic conditions are presented in Figure 2(a). The data showed that the amplitudes of A- and B-waves gradually increased with increasing stimulus intensity in all groups. Differences in B-wave amplitudes among all groups were significant at stimulation intensities of 0.01 (P = 0.005), 0.1 (P = 0.011), 1.0 (P < 0.001), and 3.0 (P = 0.014); differences in A-wave amplitudes were significant at intensities of 0.1 (P = 0.028), 1.0 (P = 0.001), and 3.0 (P = 0.002) (Figure 2(b)). A- and B-wave amplitudes of ERG responses in the Cfh+/− smoking group were markedly decreased at some stimulus intensities.
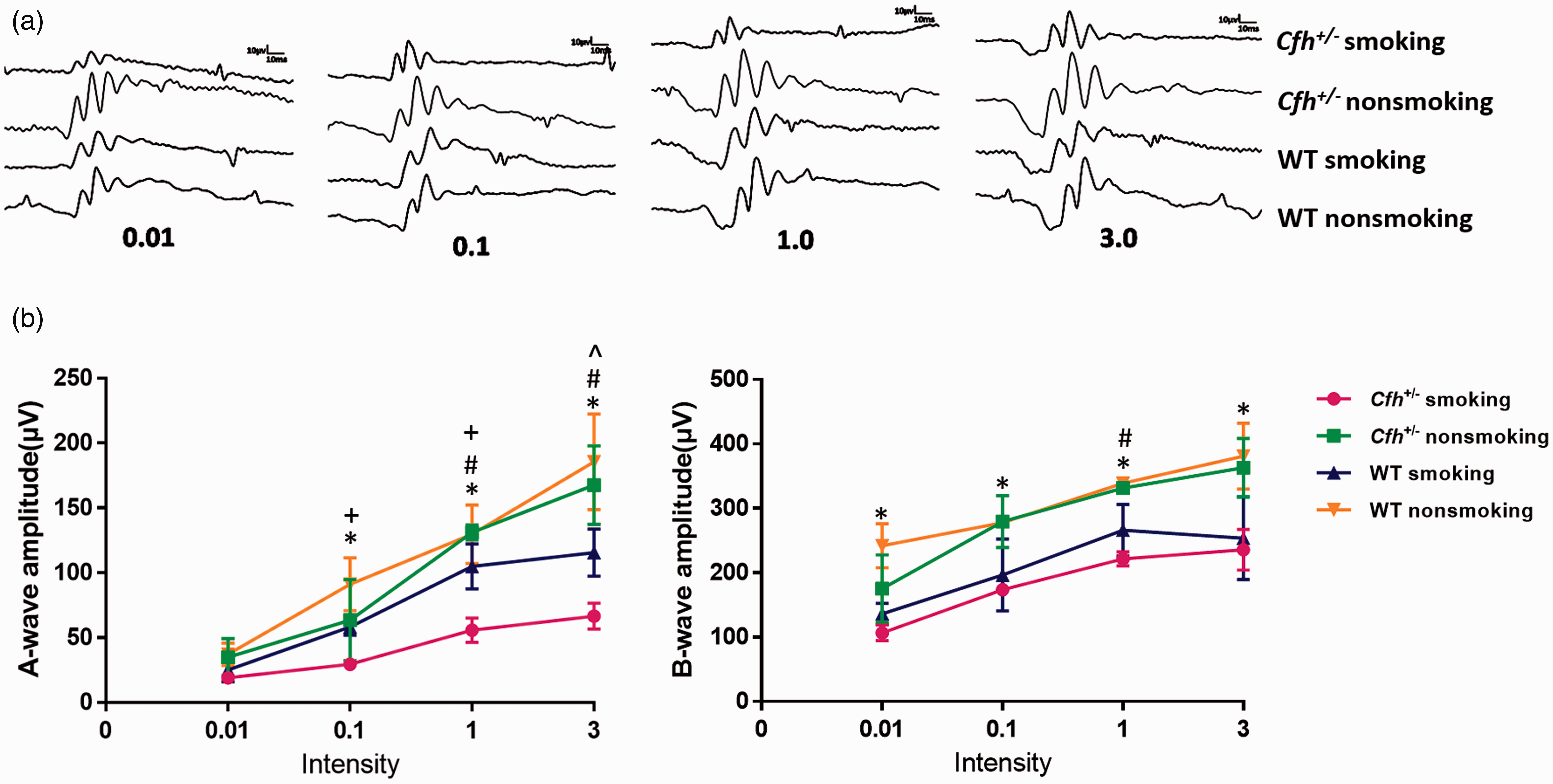
Electroretinograms of visual function in mice. (a) Amplitudes increased with flash stimuli of increasing intensity. (b) Differences in A- and B-wave amplitudes among all groups (ANOVA) (*) (n = 3). Cfh+/− smoking mice showed significantly lower A- and B-wave amplitudes than Cfh+/− non-smoking mice (#) and WT smoking mice (+). “^” means the amplitudes of WT smoking mice was significantly lower than WT non-smoking mice. (A color version of this figure is available in the online journal.)
Differences between Cfh+/− smoking and Cfh+/− non-smoking mice in A-wave amplitudes were significant at stimulation intensities of 1.0 (ANOVA, post hoc Tukey HSD, P = 0.001) and 3.0 (ANOVA, post hoc Tukey HSD, P = 0.006), while differences in B-wave amplitudes were significant at an intensity of 1.0 (ANOVA, post hoc Dunnett T3, P = 0.002) (Figure 2(b)).
Compared with WT non-smoking mice, WT smoking mice showed significantly decreased A-wave amplitudes only at a stimulation intensity of 3.0 (ANOVA, post hoc Tukey HSD, P = 0.045). Compared with WT smoking mice, Cfh+/− smoking mice showed lower A-wave amplitudes at stimulation intensities of 0.1 (ANOVA, post hoc Dunnett T3, P = 0.017) and 1.0 (ANOVA, post hoc Tukey HSD, P = 0.017) (Figure 2(b)). Since A-wave amplitude reflects rod photoreceptor function, 26 our data indicate that rod photoreceptor function is compromised in WT smoking mice and to an even greater extent in Cfh+/− smoking mice. This was further confirmed by anti-rhodopsin immunofluorescence staining which also showed the structure of photoreceptors were injured in Cfh+/− smoking mice (Figure 3).
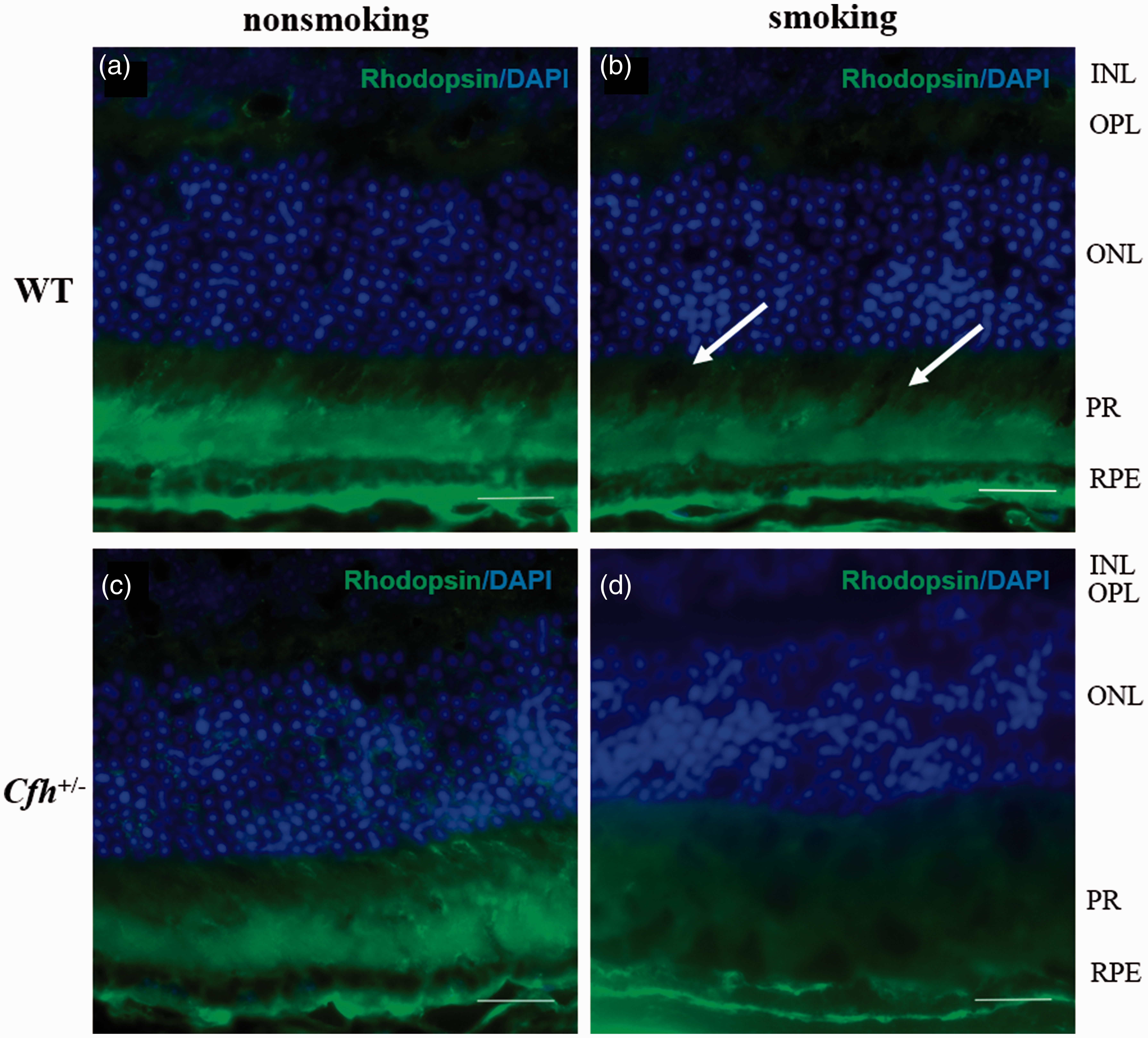
Immunofluorescence of rhodopsin. Rhodopsin staining showed the morphology and structure of photoreceptors. (a and c) Cfh+/− and WT non-smoking mice showed the intact photoreceptor structure; (b) in WT smoking mice, most of the relatively intact photoreceptor structure was preserved, with only a small amount of lesions (arrows). (d) Photoreceptor cells were not intensely labeled. Marked swelling and structural disorder in the photoreceptor of the Cfh+/− smoking mice. Scale: 20 μm. (A color version of this figure is available in the online journal.)
Smoking and CFH deficiency impaired retinal pigment epithelial cells
While fundus imaging revealed no drusen or other AMD-like pathology in Cfh+/− smoking or non-smoking mice after 12 weeks (Figure 4(a)), fundus autofluorescence (FAF) showed a large number of hyper-autofluorescence spots in a speckled pattern in Cfh+/−smoking mice (Figure 4(b)). FAF in the Cfh+/− non-smoking mice revealed normal fundus appearance (Figure 4(b)). These FAF findings indicate that RPE cells of Cfh+/− smoking mice contained significantly more lipofuscin and other metabolic waste products than RPE cells of Cfh+/− non-smoking mice.
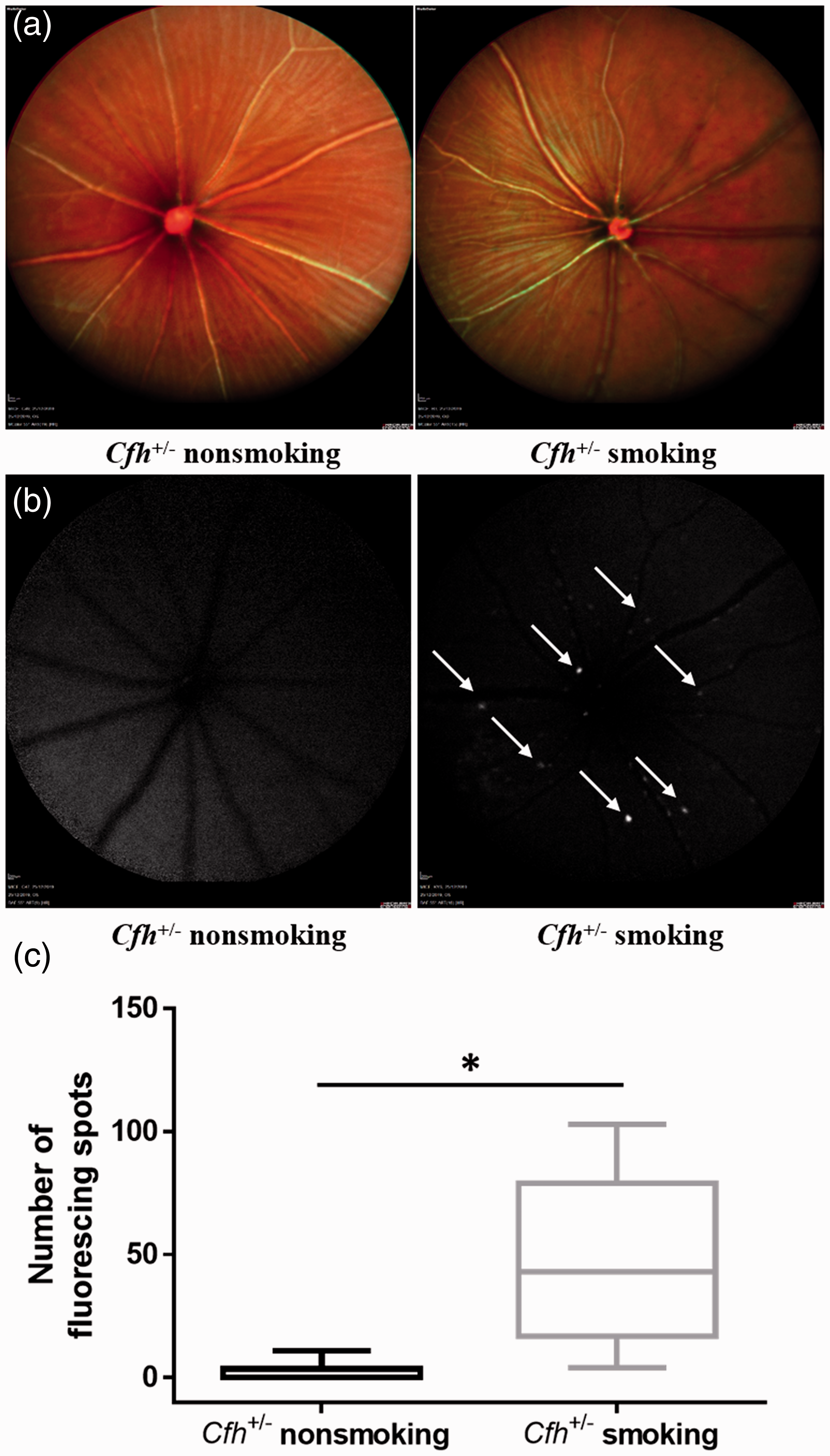
RPE cell dysfunction caused by smoking and CFH deficiency. (a) Fundus photography showed normal appearance of the fundus in Cfh+/– smoking and non-smoking mice. (b) Fundus autofluorescence (FAF) showed a large number of hyper-autofluorescence spots (arrows) in a speckled pattern in the Cfh+/– smoking mice. (c) The box plots represent the number of hyper-autofluorescence spots in the Cfh+/– smoking mice (n = 3 mice, six eyes) and Cfh+/– non-smoking mice (n = 4 mice, eight eyes). The number of hyper-autofluorescence spots was significantly greater in the Cfh+/– smoking mice (t = 3.094, P = 0.026). (A color version of this figure is available in the online journal.)
Anti-ZO-1 immunofluorescence staining of RPE-choroidal-sclera flat-mounts revealed a blurred and interrupted RPE cell tight junction in WT and Cfh+/− smoking mice (Figure 5(c) and (d)), suggesting that there was significant damage to tight junctions of RPE cells. The destruction of tight junctions was more extensive in Cfh+/− smoking mice than in WT smoking mice (Figure 5(c)). In the Cfh+/− and WT non-smoking mice, the tight junctions were intact and clearly visible between adjoining cells (Figure 5(a) and (b)).
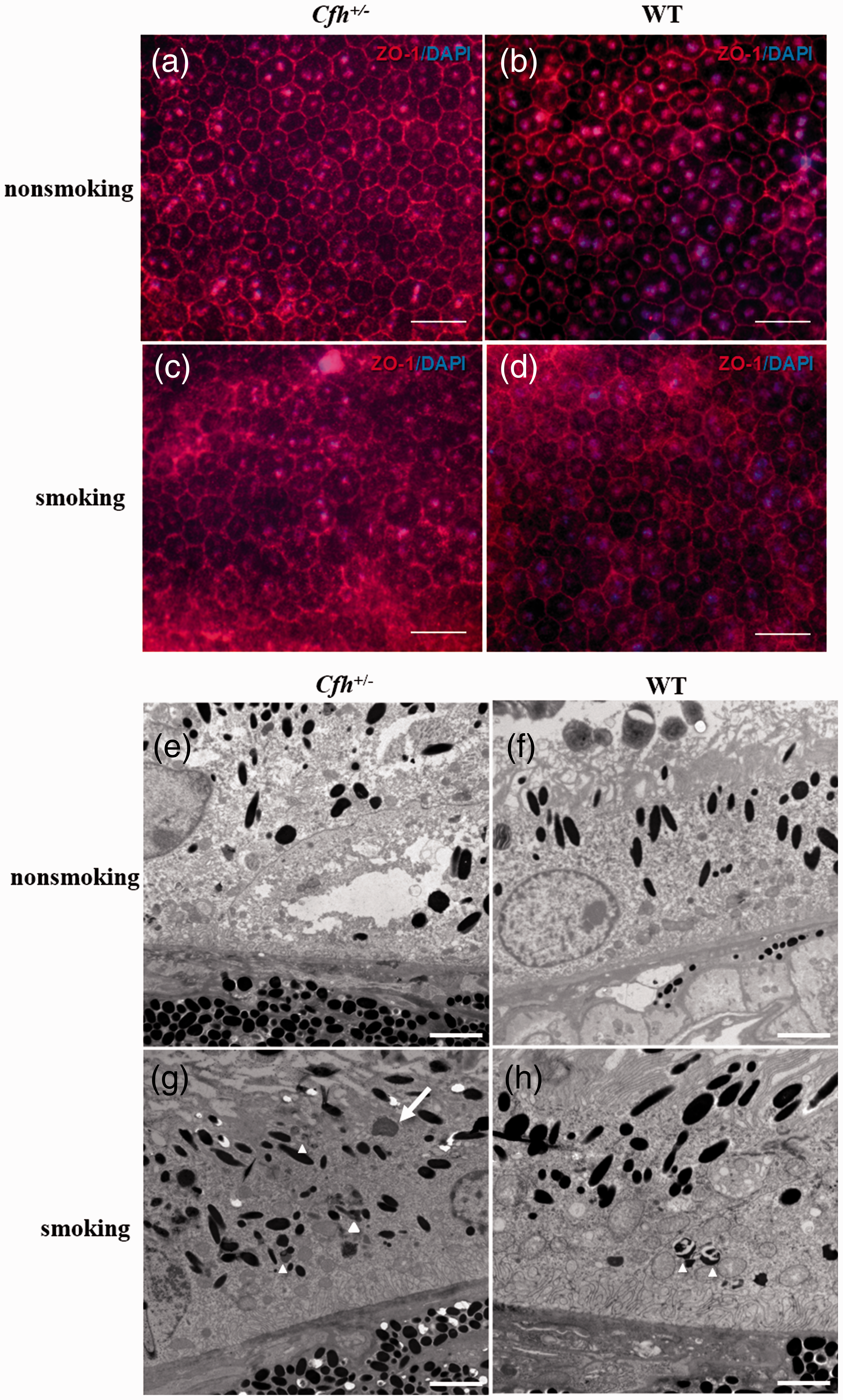
Retinal pigment epithelial (RPE) cell morphology. (a–d) Anti-ZO-1 immunofluorescent staining of RPE-choroidal-sclera flat-mounts. (c) The RPE cell tight junctions of Cfh+/– smoking mice were blurred, interrupted, and more severely damaged than those in WT smoking mice (d). (a and b) The RPE cell tight junctions in non-smoking mice were intact and clear. ZO-1 staining appears red and nuclear staining with DAPI appears blue. Scale: 20 μm. (g) The ultrastructure of RPE cells in Cfh+/– smoking mice, as revealed by transmission electron microscopy, showed undigested shed photoreceptor extracellular segments (long arrow), lipofuscin, and other particles (triangle). (h) Melanin was disintegrated in RPE cells of WT smoking mice (triangle). Scale: 2 μm. (A color version of this figure is available in the online journal.)
Transmission electron microscopy (TEM) demonstrated an accumulation of intracellular granules in RPE cells of Cfh+/− smoking mice, which might be undigested shed photoreceptor outer segments, lipofuscin particles, or other substances. Basement folds and Bruch’s membranes were generally normal (Figure 5(g)). These results indicate that smoking and CFH deficiency can lead to phagocytic and digestive dysfunction as well as structural impairment in murine RPE cells.
Smoking and CFH deficiency resulted in complement system activation and deposition of complement activation products
Optical coherence tomography (OCT) showed drusen-like accumulation at the posterior pole of the eye in Cfh+/− smoking mice, with a dome-shaped protrusion extending into the ellipsoid zone (Figure 6(a)). In contrast, Cfh+/− non-smoking mice were phenotypically normal. Immunofluorescence staining for C3b/iC3b/C3c showed normal histological findings in non-smoking WT and Cfh+/− mice (Figure 6(b) and (d)), but subretinal deposits of complement activation products in Cfh+/− and WT smoking mice (Figure 6(c) and (e)). These results suggest that smoking and CFH deficiency could hyperactivate the retinal complement system and cause deposition of complement activation products.
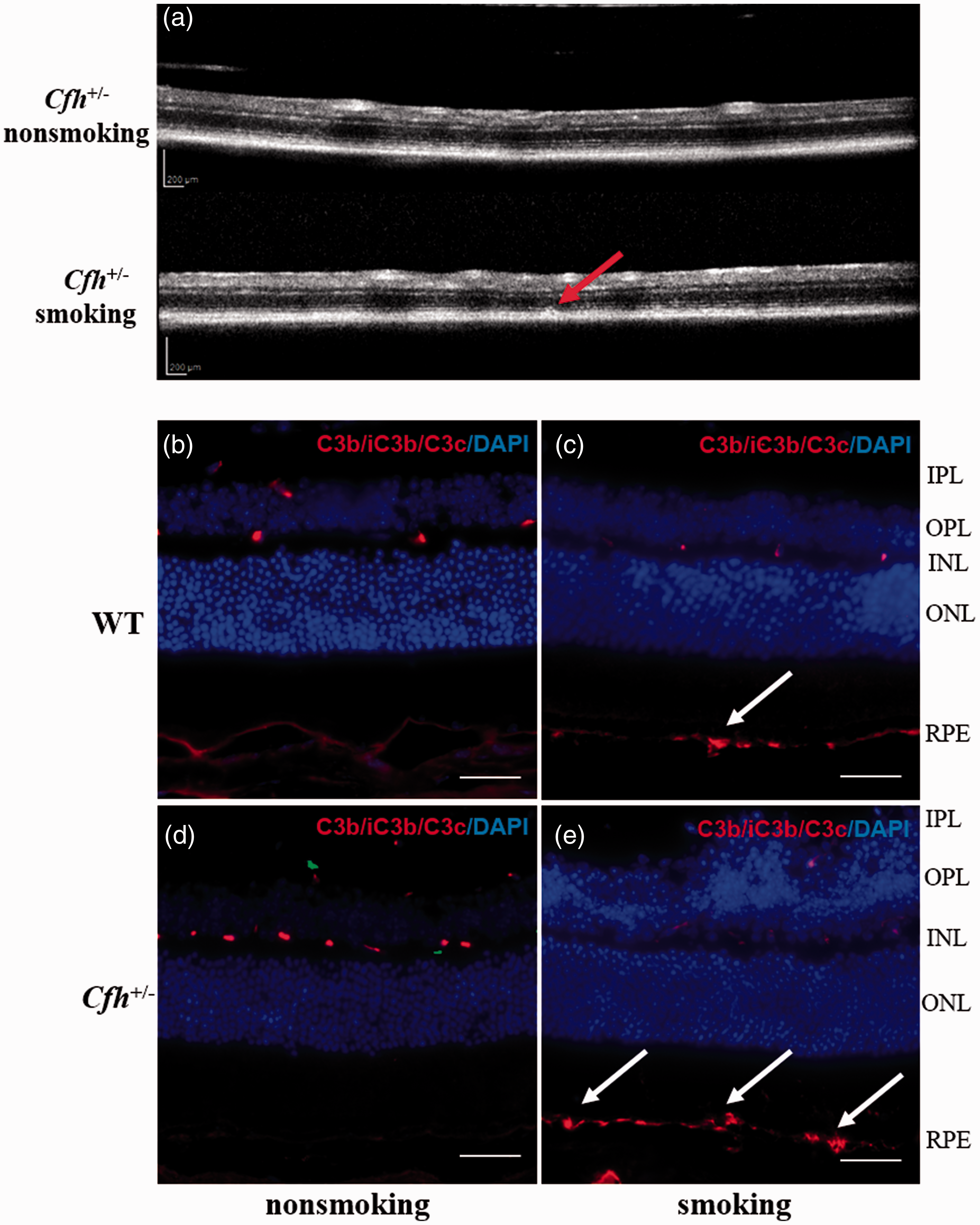
Complement system activation and deposition of complement activation products. (a) Optical coherence tomography (OCT) showed a Drusen-like accumulation in Cfh+/− smoking mice and normal morphology in non-smoking mice. (c and e) C3b/iC3b/C3c immunofluorescence staining revealed deposition of complement products in the sub-RPE of smoking mice, while C3b/iC3b/C3c were occasionally scattered in the OPL, IPL, and GCL (where the retinal vessels are located) in non-smoking animals (b and d). C3b/iC3b/C3c staining appears red, while nuclear staining by DAPI appears blue. Scale: 50 μm. (A color version of this figure is available in the online journal.)
Smoking and CFH deficiency caused retinal microglial activation
Iba1 immunofluorescence showed slight activation of microglial cells in the WT smoking mice, indicated by the dendrites of microglia extending into the outer nuclear layer. In Cfh+/− smoking mice, we detected the migration of microglia into the outer segment layer of photoreceptor cells, which is characteristic of AMD (Figure 7(d)). No microglial activation was observed in the non-smoking group (Figure 7(a) and (c)). The difference in microglial activation among the four groups was statistically significant (P < 0.001), and the post hoc Dunnett T3 test indicated that the number of activated microglial cells was significantly higher in Cfh+/− smoking mice than in Cfh+/− non-smoking (P = 0.001), WT smoking (P = 0.003), and WT non-smoking (P = 0.001) mice (Figure 7(e)). The difference in the number of activated microglial cells between WT smoking and WT non-smoking mice was statistically significant based on the independent-samples t-test (P = 0.028) (Figure 7(e)).
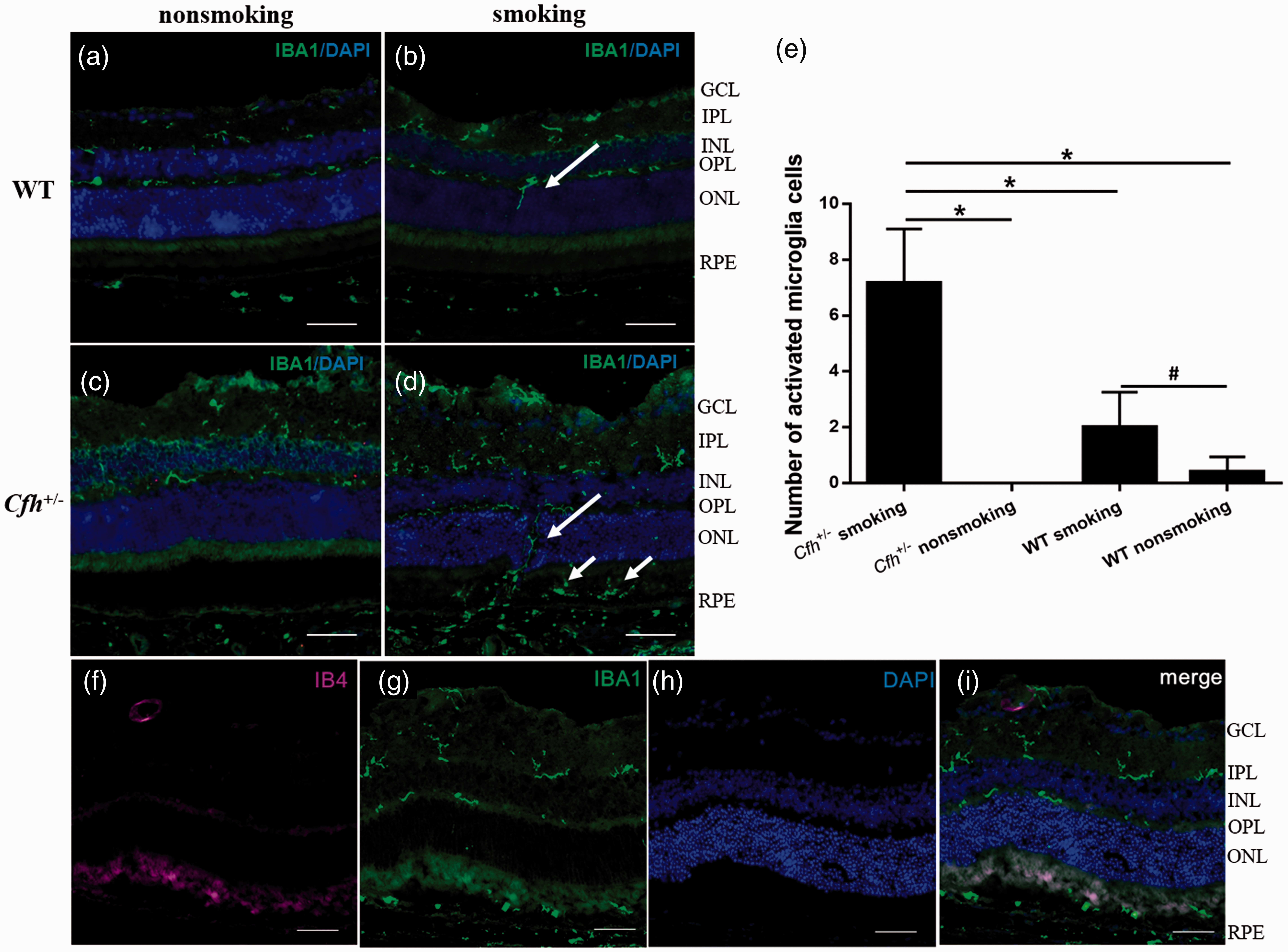
Microglial cell activation. (a and c) WT and Cfh+/− non-smoking mice showed quiescent microglia located in OPL, INL, and GCL regions. (b and d) In the WT and Cfh+/− smoking mice, the dendrites of microglia extended into the outer nuclear layer (long arrows); (d) in the Cfh+/− smoking mice, microglia migrated into the outer segment layer of photoreceptor cells (short arrows) Scale: 50 μm. (f–i) Co-staining against Iba1 and IB4 showed there was no neovascularization in the Cfh+/− smoking mice. Scale: 50 μm. (e) Quantification of activated microglial cells per field of view. Cfh+/− smoking mice showed greater microglial activation than any other group. Data are presented as mean ± SD (n = 5–6 per group). *P < 0.01 by ANOVA; #P < 0.05 by t-test. (A color version of this figure is available in the online journal.)
Retinal tissue sections from Cfh+/− smoking mice were also stained with anti-Iba1 and -IB4 antibodies to evaluate neovascularization and its spatial relationship to these activated microglial cells. There was no co-localization of the stains, suggesting that there was no neovascularization at the sites to which microglia had migrated. In fact, no neovascularization was found anywhere in the retina of Cfh+/− smoking mice (Figure 7(f) to (i)). These findings suggest that smoking can induce the activation of microglia and retinal inflammation, and that in a smoking background, abnormal activation of the complement system (i.e. in Cfh+/− mice) further activates microglia and exacerbates inflammation.
Discussion
This study provides a new animal model of AMD that may facilitate more extensive and faster preclinical studies to understand the pathology and treatment of the disease. We found that the combination of complement system over-activation and cigarette smoke exposure accelerated and strengthened the AMD-like phenotype of Cfh+/− mice, leading them to present features characteristic of early-onset AMD, including RPE cell dysfunction, deposition of complement activation products, activation of microglia, and impairment of visual function.
The complement system and AMD
The retina is an immune-privileged tissue whose blood–retina barrier protects it from damage caused by pathogenic factors. In addition to immune avoidance and tolerance, innate defense systems, including microglia and the complement system, also protect the retina. The normal function of the complement system is essential for the maintenance of the normal retinal immune microenvironment,27,28 and it plays a key role in host–defense against pathogens. However, it must be strictly regulated to avoid inflammation and tissue damage. There are three complement activation pathways: the typical, lectin, and alternative pathways. The alternative pathway is initiated by the spontaneous hydrolysis of protein C3. Activation of the alternative pathway is spontaneous and continuous. AMD involves hyperactivation of the alternative complement pathway. As a crucial inhibitor of the alternative complement pathway, CFH affects the progression of AMD by regulating oxidative stress, inflammation, and abnormal angiogenesis. 29 Moreover, the antioxidant stress function of CFH is independent of its ability to inhibit the formation of the membrane attack complex. 30
Our results showed that Cfh+/− non-smoking mice had normal retinal morphology over 20 weeks, without an AMD-like phenotype. The AMD phenotype requires a long period to develop if only driven by an abnormally activated complement system. 10 This suggests that CFH deficiency might contribute to AMD initiation and progression, but low expression of CFH alone is not sufficient to induce retinal degeneration in young mice. Alternatively, there may be some mechanisms to compensate for the CFH deficiency.
Smoking, complement activation, and AMD
A large number of studies showed that cigarette smoke could induce RPE cell apoptosis, basal laminar deposits, thickened Bruch’s membrane, impaired visual function, and other AMD-like changes in mice.14,15,31,32 In our study, we confirmed some of those changes in the WT smoking mice. The visual function of WT smoking mice was impaired, as shown by the decreased A-wave amplitudes in ERG. Under normal conditions, the RPE is one of the tissues with the highest oxygen consumption, and it is sensitive to anoxia and oxidants. Oxidants in cigarette smoke appear to underlie the pathology that harms RPE cells, 33 which is closely related to the early development of AMD. 34 It is well known that the RPE cells are part of the outer blood–retina barrier, and dysregulation of RPE integrity involves retinopathies, including AMD 35 . We found that the tight junctions between RPE cells in the WT smoking mice were destroyed, demonstrating that smoking disrupts this critical blood–retina barrier. Furthermore, we showed that the complement-activation product C3b/iC3b/C3c is deposited in the sub-RPE in WT smoking mice, which is consistent with results from other studies.36,37 This provides direct evidence that smoking can lead to complement system activation. Finally, we found that microglia in the retina were slightly activated in WT smoking mice, and that the number of activated microglia cells was significantly higher, suggesting that smoking alone can activate immune cells in mice with otherwise normal immune function. With age and in the presence of risk factors such as smoking, RPE cells change from an inflammatory state that attempts to restore homeostasis to a state of chronic inflammation associated with AMD. 38 The change in the immune microenvironment of retinas in smoking mice may initiate AMD.
Studies have shown that Cfh+/− mice at 90 weeks displayed AMD-like characteristics, while the mice under 40 weeks showed no similar changes. 10 Cfh–/– mice fed under normal feeding conditions for 96 weeks also showed AMD-like changes, 9 indicating that age plays an important role in the pathogenesis of AMD. However, Cfh–/– mice fed with low glycemic index chow showed early AMD-like characteristics at the age of 10 months (40 weeks), 39 which was more than twice as quickly as mice fed a normal diet. The authors speculated that this might be because the low-energy diet leads to oxidative stress and dysfunction of RPE cells. Under CFH deficiency, the ability of the retina to inhibit oxidative stress could then be considerably weakened, markedly accelerating the development of AMD lesions. This is similar to our findings. Our results showed that Cfh+/− mice exposed to cigarette smoke (causing oxidative stress mainly) exhibited early AMD-like changes, such as RPE cell dysfunction, accumulation of complement activation products, and activation of microglia at 20 weeks (after the 12-week intervention), and that the time to lesion occurrence was significantly shorter. Our findings suggest that the combination of CFH deficiency and smoke exposure promotes rapid AMD-like retinopathy.
As previously mentioned, the pathological changes of the retina caused by smoking are dependent on the activation of the complement system, especially the alternative complement pathway.17,40 Furthermore, inhibiting the activation of the alternative complement pathway can not only prevent but also alleviate retinal damage caused by smoking. 18 Collectively, these data suggest a close association among cigarette smoke exposure, hyperactivation of the complement system, and retinopathy.
The disruption of RPE structure and function is one of the pathologic changes of AMD. In our study, RPE cells of Cfh+/− smoking mice contained more metabolic waste (such as undigested shed photoreceptor outer segments and lipofuscin), and their tight junctions were more severely damaged than RPE cells of WT smoking mice. Both over-activation of the complement system and smoking can lead to an overload of RPE cells, resulting in dysfunction. 41 The RPE dysfunction may affect the photoreceptor metabolism and function, as demonstrated by markedly lower A- and B-wave amplitudes in the Cfh+/− smoking group than in other groups, including WT smoking mice. We hypothesize that the additional strain of CFH insufficiency weakens the ability of the complement system to protect RPE cells 42 while also hyperactivating the complement system to damage RPE cells. 43 Thus, CFH insufficiency renders RPE cells more susceptible to smoke-induced damage.
Increased autophagy in RPE cells and CFH deficiency have been associated with the formation of depositions, and may contribute to the formation of drusen.44,45 In our study, Cfh+/− smoking mice showed accumulation of complement activation products C3b/iC3b/C3c, and OCT revealed drusen-like deposition in the same location. This may be caused by the deficiency of CFH and/or the dysfunction of RPE cells.
Microglial activation and complement activation
The immune cells of the retina are composed entirely or almost entirely of microglial cells 46 , which play an important role in regulating the inflammatory response. Resting retinal microglia are located in the ganglion cell layer, the inner plexus layer, and the outer plexus layer, while the outer nuclear layer is devoid of microglia. In pathological conditions such as AMD, diabetic retinopathy, and glaucoma, microglia are found in the outer nuclear layer and subretinal region.47,48 We also observed activated microglia cells in Cfh+/− smoking and WT smoking mice. Microglia activation involves changes in morphology and distribution: 49 microglia transform from a ramified morphology to a hypertrophied or elongated fusiform morphology and migrate to the lesion. Our results showed that the retinal microglial cells in the WT smoking mice were slightly activated, with only dendrites extending into the outer nuclear layer. In the Cfh+/− smoking mice, microglia were very active, with dendrites reaching the sub-RPE and a large number of microglia migrating to the photoreceptor outer segment layer. Our model shows a phenotype consistent with that of other animal models of early AMD, 10 and it supports the idea that microglial activation contributes to the early pathogenesis of several retinal disorders, including AMD.47,50
Abnormal activation of the complement system can directly damage retinal tissue and recruit immune cells into the vicinity of the complement cascade reaction, leading to the infiltration of microglia and chronic inflammation. Inhibition of complement activation pathways in mice dampens the inflammatory response. 51 Activation of the complement system has a certain influence on the activation of microglial cells. 52 In our study, while no significant differences in C3b/iC3b/C3c deposition (a marker for complement activation) were found between WT and Cfh+/− smoking mice, microglia activation was higher in the Cfh+/− smoking mice than in WT smoking mice. Further study should examine the initiators and pathways of microglial activation in our Cfh+/− smoking animals. Specifically, experiments should explore whether there are differences between Cfh+/− and WT smoking mice in the whole complement reaction and the reason of different activation of microglia cell between WT and Cfh+/− smoking mice.
In this study, an early AMD murine model was successfully established by combining smoking exposure and complement over-activation for the first time. The new method greatly shortens the time to achieve the relevant disease phenotype. Future work should observe the animals over longer periods and experiment with different ages at initial exposure, and it may be possible to induce a phenotype that mimics late AMD. And further experiments are needed to elucidate the specific mechanisms underlying the pathological changes of this model.
Supplemental Material
sj-pdf-1-ebm-10.1177_15353702211052245 - Supplemental material for Complement factor H deficiency combined with smoking promotes retinal degeneration in a novel mouse model
Supplemental material, sj-pdf-1-ebm-10.1177_15353702211052245 for Complement factor H deficiency combined with smoking promotes retinal degeneration in a novel mouse model by Liwen Feng, Kailai Nie, Qing Huang and Wei Fan in Experimental Biology and Medicine
Footnotes
AUTHORS’ CONTRIBUTIONS
All authors participated in the design, interpretation of the studies and analysis of the data and review of the article; LF and KN conducted the experiments, LF wrote and edited the article, and WF edited the article.
ACKNOWLEDGMENTS
We would like to thank Professor Tao Wang and Dr. Jun Chen from the Respiratory Laboratory of Sichuan University for providing smoking equipment and guidance for animal models. We appreciate the assistance of Professor Song Lei of Sichuan University for technical support with electron microscopy. We thank Professor Danian Chen of Sichuan University for providing us with anti-IB4 antibody.
DECLARATION OF CONFLICTING INTERESTS
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
FUNDING
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This work was supported by The National Natural Science Foundation of China (81670869 to Wei Fan).
SupplementAL MATERIAL
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.