
Abstract
LncRNA expression can be conducive to gastric cancer (GC) prognosis. The objective of this study is to ascertain five specific lncRNAs involved in tumor progression of GC and their role as prognostic markers to diagnose clinical stage-wise GC. High-throughput RNA sequencing data were obtained from The Cancer Genome Atlas (TCGA) database and performed genome-wide lncRNA expression analysis using edgeR package, Bioconductor.org, and R-statistical computing to analyze differentially expressed lncRNA analysis. Cutoff parameters were FDR < 0.05 and |Log2FC| > 2. Total 351 tumor samples with differentially expressed lncRNAs were divided into group-1 lncRNAs such as AC019117.2 and LINC00941, and group-2 lncRNAs such as LINC02410, AC012317.2, and AC141273.1 by 2:1. The Spearman correlation coefficients (p < 0.05) and correlation test function (cor.test ()) were performed for lncRNAs as per clinical stage. Cytoscape software was used to construct lncRNA–mRNA interaction networks. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway (p < 0.05) analysis were conducted using the clusterProfiler package. Kaplan–Meier survival analysis was performed to determine the overall survival of patients based on the expression of five lncRNAs in different clinical stages of GC. AC019117.2 and LINC00941 of group 1 inferred a positive correlation with clinical stages of stage I to stage IV, and their expressions were higher in tumor tissues than normal tissues. On the contrary, LINC02410, AC012317.2, and AC141273.1 of group 2 exhibited a negative correlation with clinical stage, and they exhibited more expression in normal tissues compared to tumor tissues. GO and KEGG pathway analysis reported that AC019117.2 may interact with LINC00941 via ITGA3 and trophoblast glycoprotein (TPBG) to foster tumor progression. Tumor-specific group-1 lncRNAs were conducive to the poor overall survival and exhibited a positive correlation with the clinical stages of stage I to stage IV in GC as per the lncRNA–mRNA networking analysis. These five lncRNAs could be considered as clinically useful lncRNA-based prognostic markers to predict clinical stage-wise GC progression.
Impact Statement
LncRNA expression may exhibit a significant role for effective gastric cancer (GC) prognosis based on the clinical stage-wise GC. Using the systematic integrating bioinformatics of The Cancer Genome Atlas (TCGA) database for GC models, the current study analyzed five lncRNAs signature in tumor progression of GC. LINC00941 is positively correlated with the clinical stage of the tumor, and its higher expression has a significant relationship with poor prognosis. AC019117.2 and LINC00941 inferred a positive correlation with clinical stage, and their expression was higher in tumor tissues than normal tissues. On the contrary, LINC02410, AC012317.2, and AC141273.1 exhibited negative correlation with clinical stage, and their expression was higher in normal tissues compared to tumor tissues. These lncRNAs were identified pertaining to the clinical stage through TCGA database, and could be used to distinguish the malignancy of GC patients and further provide a possible way to the early prognosis and early personalized treatment of GC.
Introduction
Gastric cancer (GC) is the fifth most commonly diagnosed cancer and has become a vital public health burden in different nations. 1 Several therapeutic modalities against GC such as surgery, chemotherapy, Helicobacter pylori treatment, and radiotherapy were developed; especially the neoadjuvant chemotherapy has been preferred for treating the advanced-stage GC. 2 , 3 The clinical stage during diagnosis can elucidate the prognosis of patients with GC. For instance, the patients with localized, early-stage GC have been reported to exhibit >60% overall survival (OS) rate, but the five-year OS rates for GC patients with local and distant metastases typically decrease to 30% and 5%, respectively. 4 However, the five-year OS rate pertinent to GC remains unsatisfactory despite diagnostic and therapeutic advances due to the debilitating progression of advanced disease stages when patients are diagnosed, especially in China. 5 Therefore, exploring the novel biomarkers for predicting GC prognosis remains an important strategy.
Recent advancements in high-throughput sequencing technologies have provided many benefits in terms of screening prognostic genes in the transcriptomic data for advanced-stage cancers. Research mining of The Cancer Genome Atlas (TCGA) and gene expression omnibus (GEO) databases delineated lncRNAs as potential prognostic factors for GC patients as tumor suppressor genes or oncogenes, and they could regulate tumor cell biological processes. 6 LncRNAs exhibit potential ability to modulate gene expression at transcriptional and post-transcriptional levels.7–9 Furthermore, divergent expressions of lncRNAs can modulate cancer development, progression 10 , 11 by fostering several oncogenic signaling to enhance tumor cell proliferation, anti-apoptosis, and metastasis, whereas some of lncRNAs reported to be implicated in diagnosis and prognostication.12–17
Several prognostic biomarkers including fibroblast growth factor receptor, 18 human epidermal growth factor receptor 2 (HER2), 19 epidermal growth factor receptor (EGFR), 20 and hepatocyte growth factor receptor (HGFR) 21 have been undergoing clinical trials for predicting GC. However, these biomarkers are associated with accuracy and reliability limitations to promote them as potential biomarkers. Divergent expression of lncRNAs through TCGA data mining for GC could be a beneficial strategy to improve prevention, and early diagnosis and treatment against various cancers. 22 Hence, it is crucial to discover and identify novel lncRNA-based signatures with high robustness and reliability to enhance clinical outcomes in the patients through the stage-wise prognosis of GC.23–25 Extensive cross-validation of these lncRNA-based predictive signatures have been required in several cohorts. 6
The lnc-TALC can confer temozolomide resistance invoked through Akt signaling in glioblastoma (GBM) cells. 26 Furthermore, lnc-SChLAP1 expression fosters the GBM tumor cells proliferation and growth via ACTN4 stabilization and by promoting activity of NF-κB signaling. 27 LncRNAs including “LNC01545, WDR11-AS1, NDUFA6-DT, FRY-AS1, and TBX5-AS1” are reported to be implicated in OS of GBM patients suggesting their significant role as biomarkers to diagnose GBM. 28 Expression patterns of lncRNAs including “UCA1, MALAT1, HOXA11 (HOXA11 antisense RNA)-AS, and ZEB1-AS1”29–31 can be used as diagnostic or prognostic markers for GC, but their mechanism by which these non-coding RNAs regulate GC has yet to be determined. Another study by Xianqin Zhang et al. 32 confirmed the implications of lncRNAs including UCA1, HOTTIP, and HMGA1P4 in the prognosis of GC. Zeng et al. 33 suggested a positive correlation of LINC00675 with the prognosis by enhancing phosphorylation of vimentin to suppress GC development. Another report delineated the functional role of LINC00675 in regulating cancer cell proliferation, migration, and invasion via Wnt/β-catenin signaling. 34 Zhao et al. 35 concluded that Plasmacytoma variant translocation 1’ (PVT1)-mediated angiogenesis by activating the STAT3/VEGF-A signaling axis, further promoting tumor growth. Another report by Song et al. 36 examined the clinical data pertinent to the patients with or without lymph node metastasis in GC and performed lncRNA expression profiles. This study revealed that the high-regulated XLOC_010235 and low-regulated RP11-789C1.1 exhibited a positive correlation to the lymph node, local and distant metastases, and TNM (tumor, node, metastasis) stage, and subsequently determined their roles as poor prognostic biomarkers in GC. 36
Thus, the clinical role of lncRNAs expressed as per clinical stage and pathological grade has been confirmed in gliomas, 37 ovarian cancer, 38 osteosarcoma, 39 clear cell renal cell cancer, 40 bladder cancer, 41 and cervical cancer. 42 Functional enrichment analysis by Haiming Liu et al. 43 suggested that LINC00941 oncogene co-expression network can regulate GC tumor cell proliferation and metastasis. Loss-of-function studies of this gene significantly impaired cancer cell proliferation, migration, and invasion, and consequently modulated GC tumor growth. 43 However, there are very limited reports pertaining to the lncRNA transcriptome data analysis to screen clinical stage-wise and pathological grade-wise biomarkers for GC patients. In this study, we employed bioinformatics to screen lncRNA biomarkers related to the different tumor clinical stages including stage I to stage IV by ascertaining transcriptome profiles and clinical data mining of GC from TCGA database; our study identified five clinical stage-wise expressions of lncRNA biomarkers according to our pipeline. Then, we performed correlation analysis between these five lncRNAs and mRNAs, and subsequently established the lncRNA–mRNA co-expression network to find out the interaction between AC019117.2 and LINC00941 in tumor progression of GC. Finally, we performed the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and gene ontology (GO) analysis to understand the underlying mechanisms of the five lncRNAs.
Materials and methods
Database search and data collection
Level 3 RNA-Seq gene expression profile of GC patients was downloaded from the TCGA database (https://portal.gdc.cancer.gov/). Meanwhile, the relevant clinical data were also downloaded from the TCGA data portal. We excluded samples without a clear clinical stage, and further screened their expression patterns of lncRNAs and mRNAs, respectively, according to the GENCODE database (https://www.gencodegenes.org/).
All data preprocessing and processing were executed according to the methodology described by Xie et al. 44 The source code will be provided in supplementary files upon request as Code R and STAD.lnc.tumor.stage file for better understating the methodology with more clarity.
Differentially expressed lncRNA analysis
In order to obtain differentially expressed lncRNAs in cancer tissue as well as normal tissues, primarily several lncRNAs whose count value was 0 were eliminated. Subsequently, the edgeR package, Bioconductor.org, and R-statistical computing were employed to perform the differentially expressed lncRNA analysis. The cutoff parameters were FDR < 0.05 and |Log2FC| > 2.
Identification of lncRNAs with clinical stage
The lncRNAs were obtained from 382 samples (31 normal samples and 351 tumor samples). Total 351 tumor samples with differentially expressed lncRNAs were randomly divided into groups as follows: group 1 and group 2 by 2:1. The Spearman correlation coefficients (p < 0.05) for lncRNAs and clinical stage were separately calculated using correlation test function (cor.test()) in R software. We screened group-1 lncRNAs and group-2 lncRNAs accurately. Group-1 lncRNAs exhibited a positive correlation with clinical stage, and their expression profile was reported to be decreased in normal tissues than tumor tissues; group-2 lncRNAs exhibited a negative correlation with clinical stage, and their expression profile was higher in normal tissues than tumor tissues. The above analysis was repeated 100 times for avoiding the random allocation bias. The above two groups of lncRNAs with consistent correlation were selected for further analysis.
lncRNA–mRNA interaction analysis, GO, and KEGG pathway analysis
Interaction analysis of lncRNAs and mRNAs was performed using cor.test() in R-statistical computing to obtain the selected lncRNAs and their correlated target genes (r > 0.53 and p < 0.05). Cytoscape software was used to elucidate lncRNA–mRNA interaction networks. GO process and KEGG pathway (p < 0.05) analysis were performed to explore biological features of selected lncRNAs and their correlated genes using the clusterProfiler package (Bioconductor.org).
Kaplan–Meier survival analysis
Kaplan–Meier survival analysis was executed with the aid of R-statistical computing, and comparisons were performed by log-rank test to elucidate prognostic significance of selected lncRNAs. Level of significance was defined as p value <0.05.
Results
Differentially expressed lncRNAs between normal and tumor samples
Total 14,776 lncRNAs were obtained from 382 samples (31 normal samples and 351 tumor samples). Subsequently, a total of 636 lncRNAs were screened using edgeR package in R-statistical computing; among them 529 lncRNAs were upregulated whereas 107 lncRNAs were downregulated. A volcano plot was constructed to discern a significant difference between two groups of differentially expressed lncRNAs (Figure 1(a)). Top 50 lncRNAs were shown in heatmap and principal components analysis (PCA; Figure 1(b) and (c)).
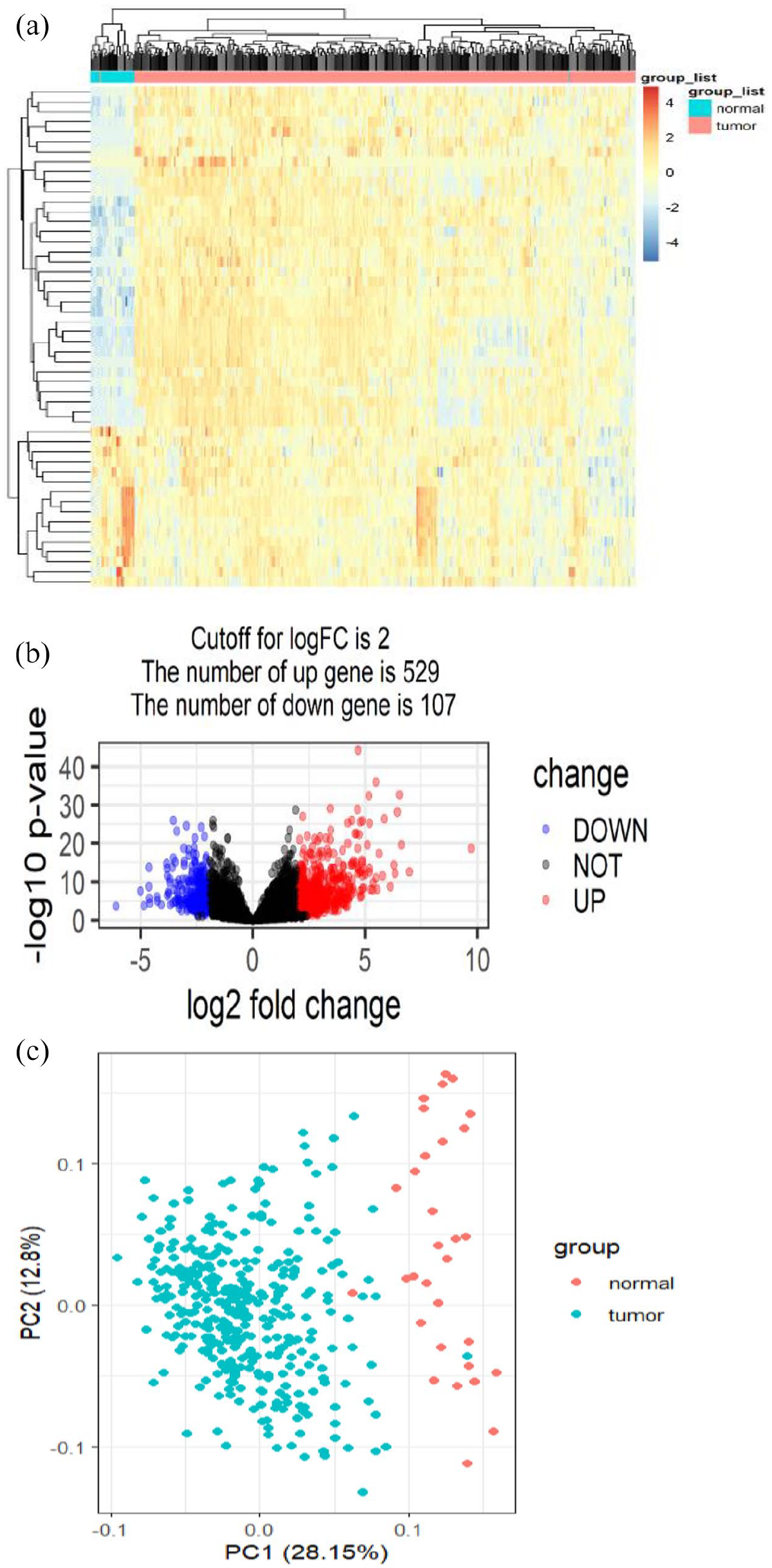
The differentially expressed lncRNAs were analyzed with the aid of edgeR package in R-statistical computing. (a) Volcano plot showed the number of differentially expressed lncRNAs (FDR < 0.05 and |Log2FC| > 2). (b) Top 50 differentially expressed lncRNAs were depicted in the heatmap. (c) Top 50 differentially expressed lncRNAs were depicted in principal components analysis (PCA). (A color version of this figure is available in the online journal.)
lncRNAs with clinical stage and pathological grade of GC
According to our pipeline, five lncRNAs met the criteria after repeating the analysis for 100 times. Among them, two lncRNAs such as AC019117.2 and LINC00941 are considered as group 1, whereas three lncRNAs such as LINC02410, AC012317.2, and AC141273.1 are considered as group 2. Group-1 lncRNAs exhibited a positive correlation with the clinical stage, and their expression was higher in the tumor tissues when compared to normal tissues. On the contrary, group-2 lncRNAs exhibited a negative correlation with pathological clinical stage, and their expression was typically higher in normal tissues when compared to tumor tissues (Figure 2(a)). The frequency of both groups of lncRNA expressions was shown in Figure 2(b). Furthermore, the expression of group-1 lncRNAs AC019117.2 and LINC00941 was decreased in the normal samples and increased with the clinical stage of stage I to stage IV in tumor samples (Figure 3(a) and (b)). The expression of group-2 lncRNAs such as LINC02410, AC012317.2, and AC141273.1 was significantly higher in the normal samples and comparatively lesser in tumor samples of clinical stage I to stage IV (Figure 3(c) to (e)). LINC00941, also referred to as lncRNA-MUF, involved in mediating invasion depth, lymphatic metastasis, and TNM stage. 45 This report has concluded that LINC00941 may contribute to poor tumor difference and further testified that our results were credible.
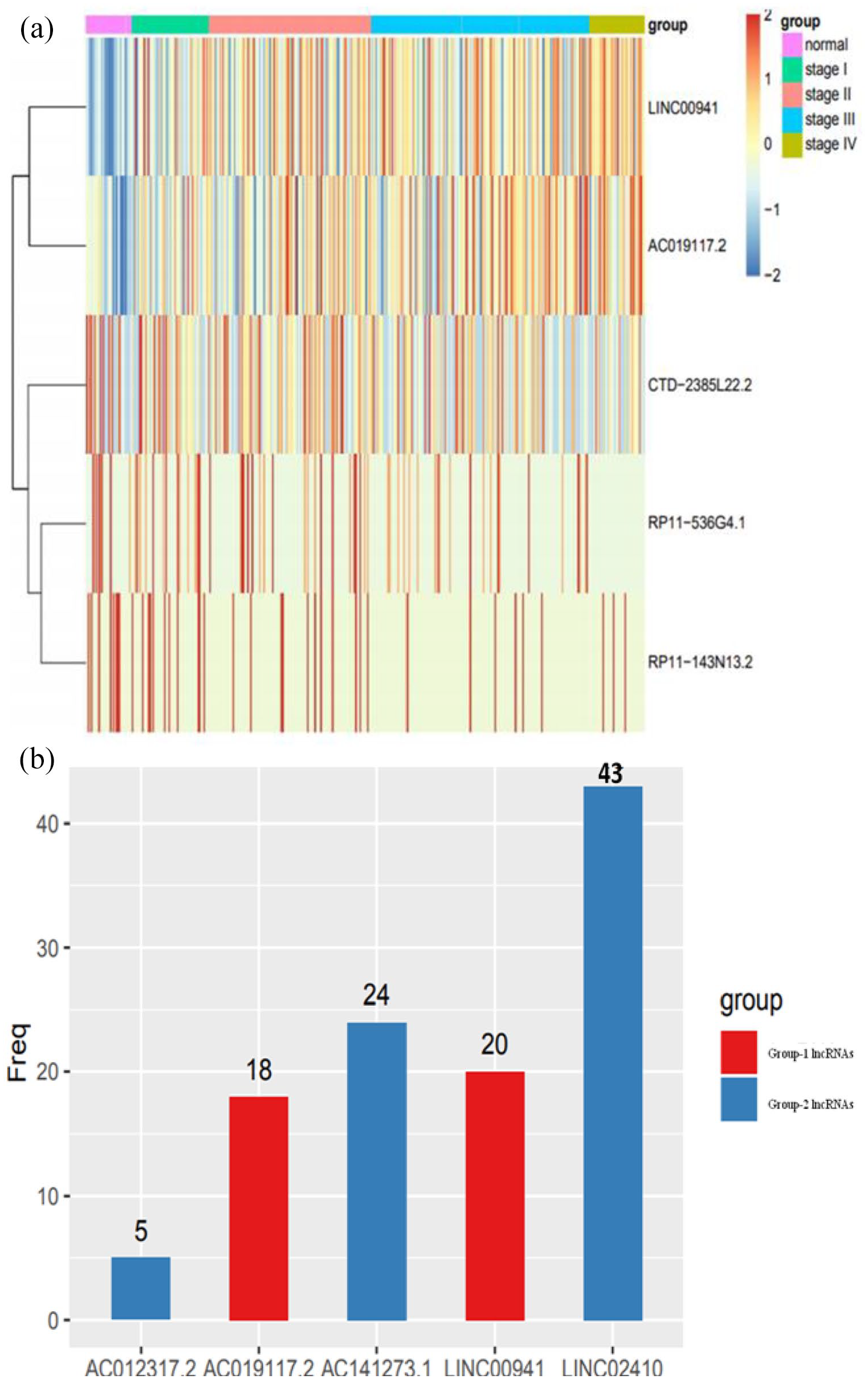
(a) Heatmap shows the expression of lncRNAs in group-1 lncRNAs (AC019117.2 and LINC00941) and group-2 lncRNAs (LINC02410, AC012317.2, and AC141273.1) in normal and different clinical stages (stage I to stage IV) of GC tumor tissue. (b) The frequency of these five lncRNAs was represented in the histogram randomly after repeating 100 times. (A color version of this figure is available in the online journal.)
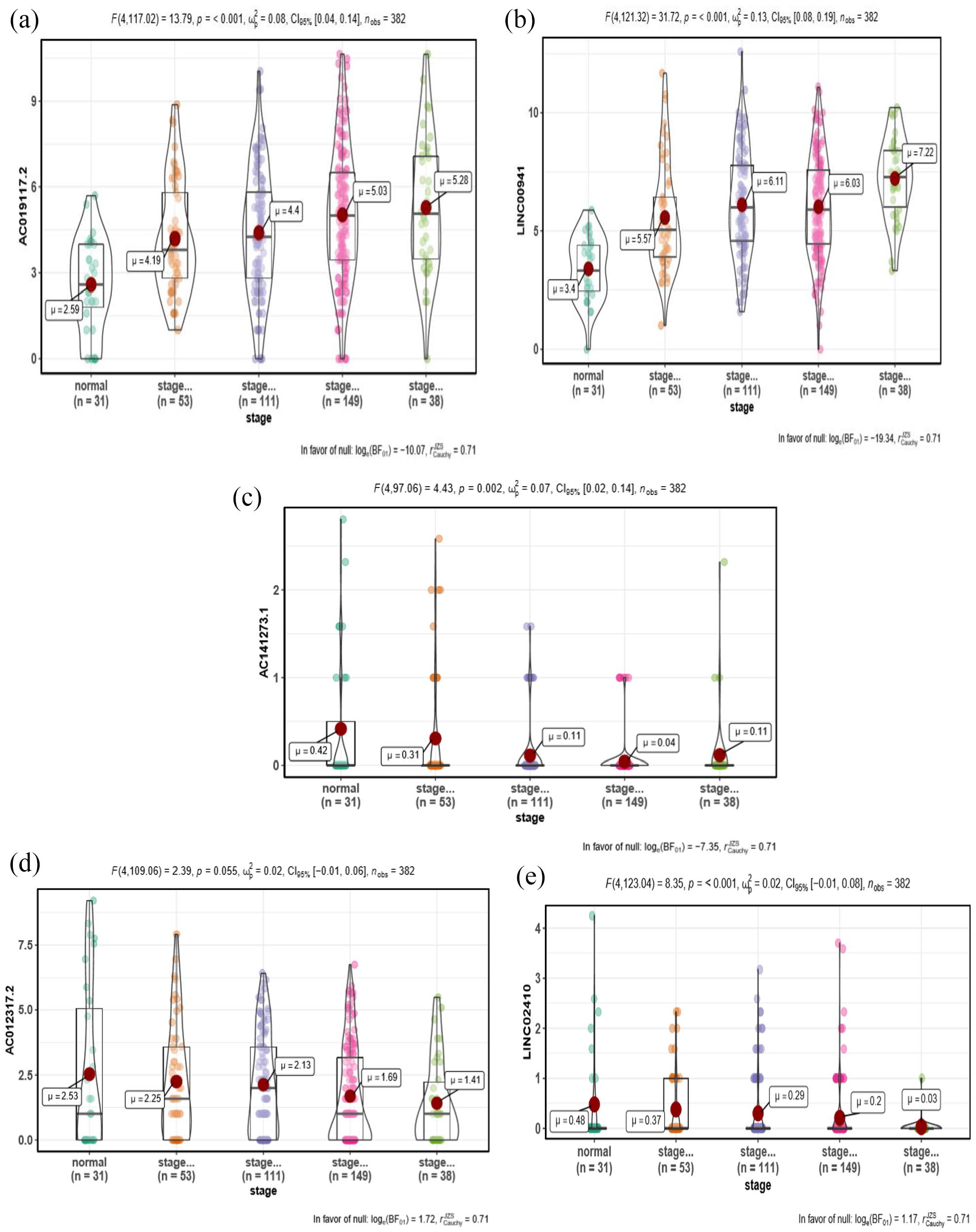
(a, b) Box plot represents that lncRNAs in group-1 lncRNAs (LINC00941 and AC019117.2) were decreased in the normal samples and increased with the clinical stages of stage I to stage IV in tumor samples, and exhibited a positive correlation. (c) to (e) Box plot represents that lncRNAs in group-2 lncRNAs (AC141273.1, AC012317.2, and LINC02410) were increased in the normal samples and decreased with the clinical stage I to IV in tumor samples, and exhibited a negative correlation. (A color version of this figure is available in the online journal.)
lncRNA and mRNA interaction analysis
The Spearman correlation statistical method was used to conduct IncRNA and mRNA interaction analysis and subsequently identified the mRNAs that were closely related to these five lncRNAs (r > 0.03 and p < 0.05). A significant correlation was observed between LINC00941 and AC019117.2 (Figure 4). Then, the lncRNA and mRNA interaction network was obtained and represented in Figure 5. Integrin subunit alpha-3 (ITGA3) and trophoblast glycoprotein (TPBG) were reported to be connecting AC019117.2 and LINC00941 in the network as they were significantly involved in tumor cell cycle, invasion, proliferation, and metastasis. 46 , 47 AC019117.2 may interact with LINC00941 via ITGA3 and TPBG to exert its role in the tumor cell biological process. This network interaction significantly demonstrates the involvement of five lncRNAs in the progression of GC.
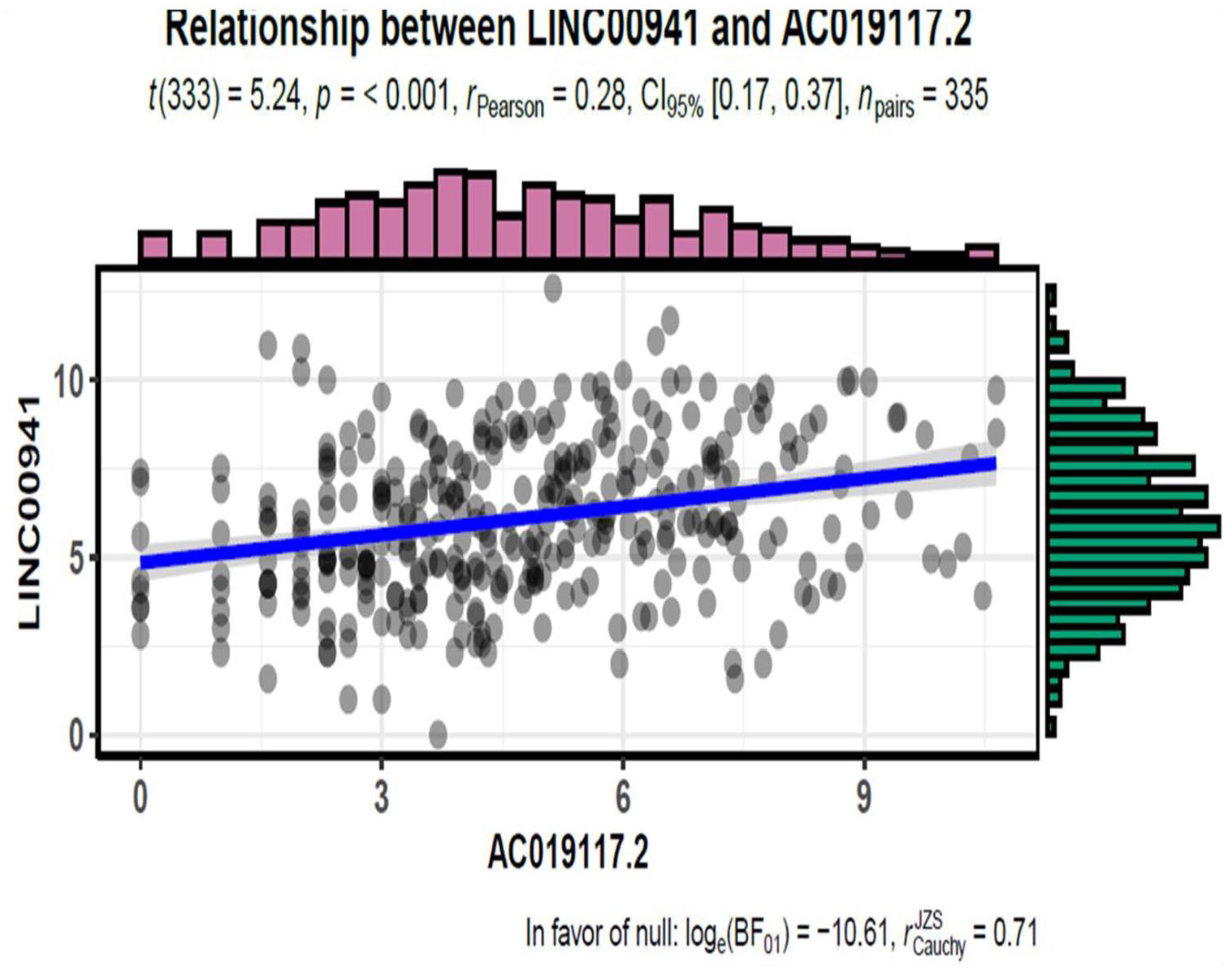
A significant correlation was observed between LINC00941 and AC019117.2 (p < 0.001). (A color version of this figure is available in the online journal.)
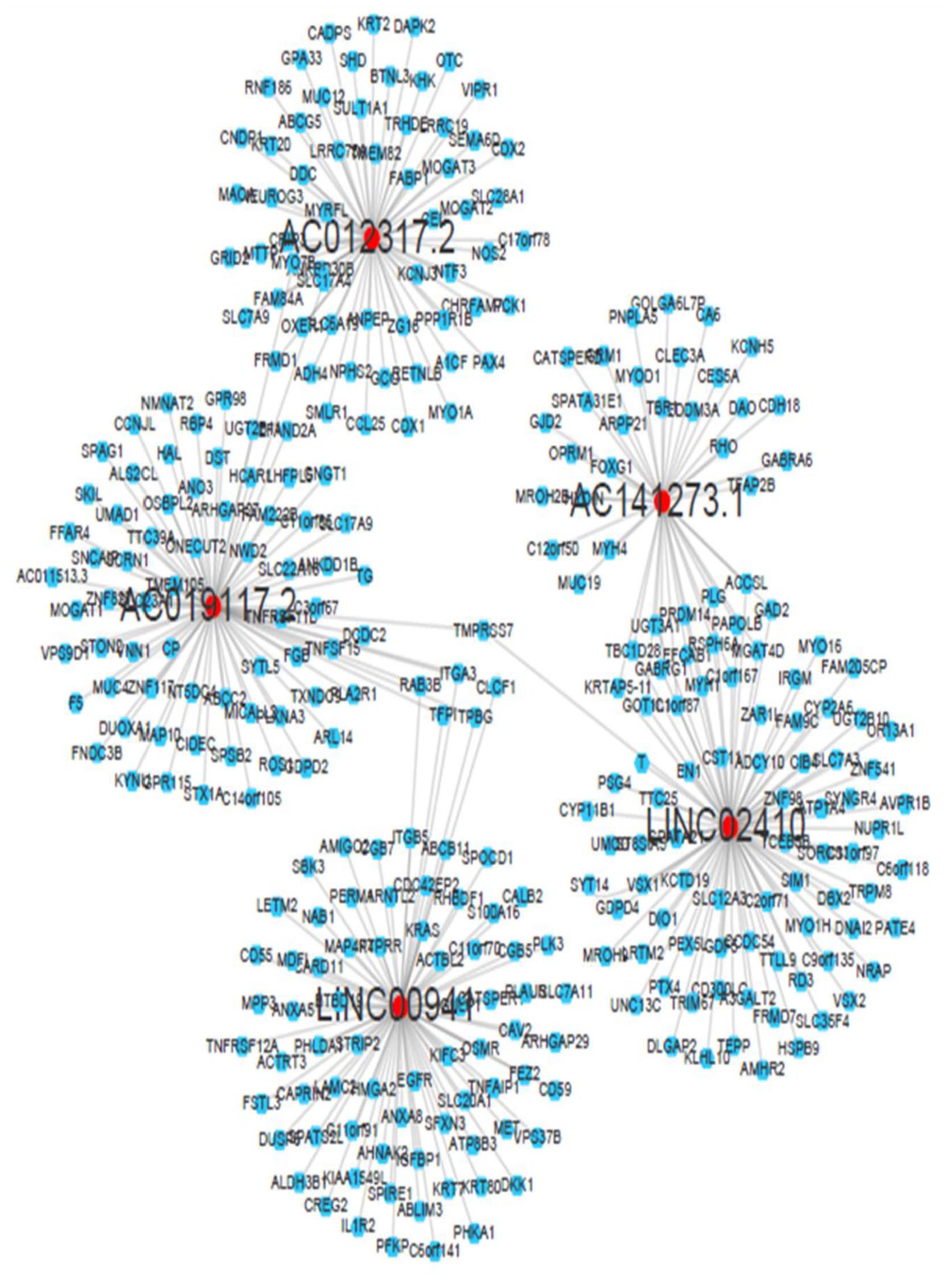
lncRNA and mRNA interaction network for these five lncRNAs (r > 0.5 and p < 0.05). Red color represents lncRNA, and blue color represents target mRNA. ITGA3 and TPBG connect AC019117.2 and LINC00941 in the lncRNA–mRNA network as these lncRNA-controlled mRNAs could significantly regulate tumor cell cycle, invasion, proliferation, and metastasis. (A color version of this figure is available in the online journal.)
Prognostic value of five lncRNAs in OS of GC patients
Kaplan–Meier curves were constructed to ascertain OS, and subsequently we reported the potential prognostic value of these five lncRNAs (Figure 6). Among indicated five lncRNAs, the expression of AC019117.2 and LINC00941 (group-1 lncRNAs) was conducive to the poor OS; furthermore, the increased expression of these two lncRNAs was positively correlated with clinical stages of stage I to stage IV – this observation was exactly consistent with the fact that higher the clinical stage, the worse the prognosis. These results were in line with previous studies. 48 Our results also delineated that three lncRNAs of group-2 lncRNAs such as LINC02410, AC012317.2, and AC141273.1 expressions were negatively correlated with clinical stages of stage I to stage IV in GC patients.
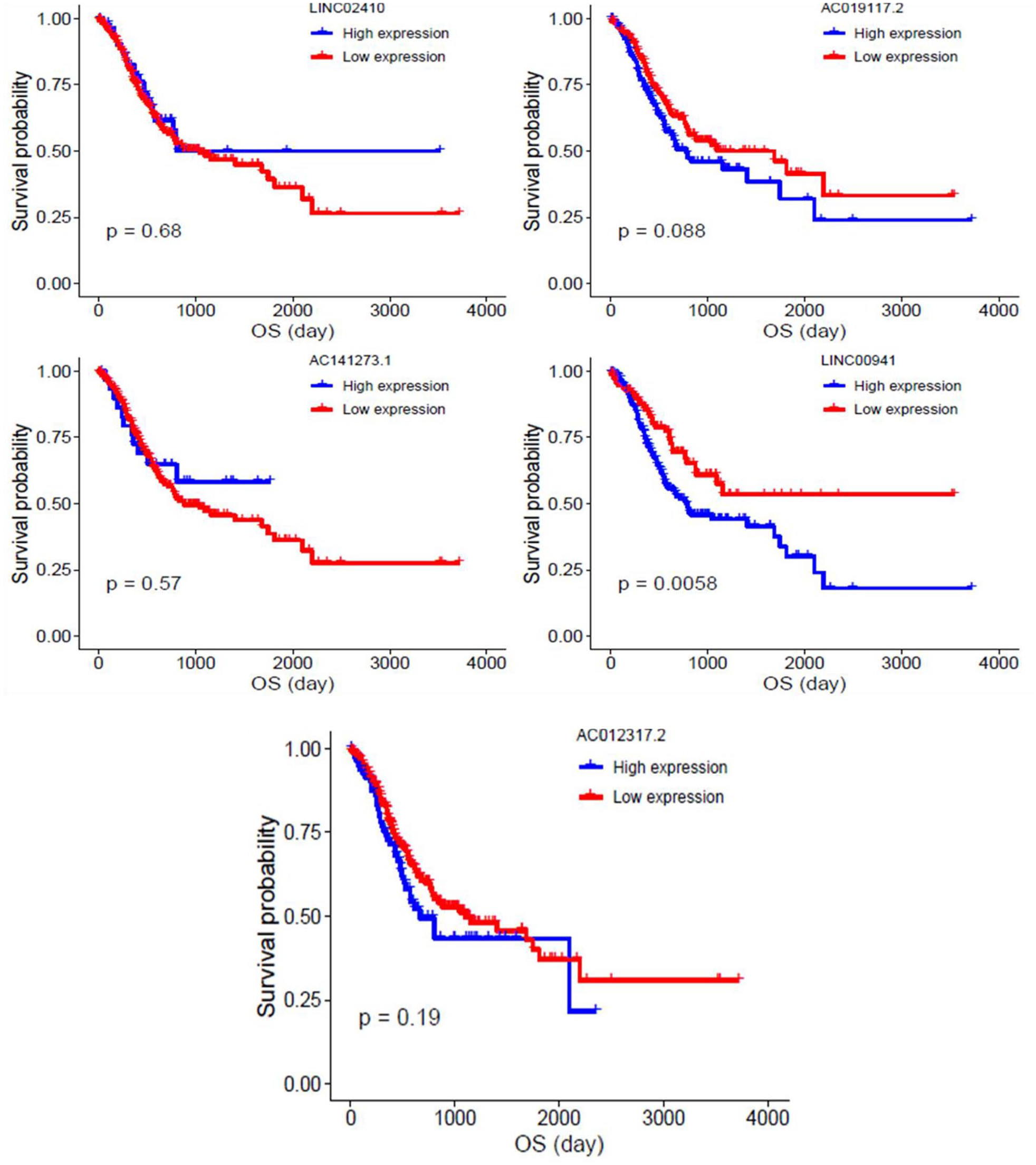
Survival analysis for these five lncRNAs (AC141273.1, AC012317.2, LINC02410, AC019117.2, and LINC00941). Extensive expression of group-1 lncRNAs (AC019117.2 and LINC00941) was reported to cause poor overall survival in GC patients. (A color version of this figure is available in the online journal.)
GO and KEGG pathway analysis
The function of two lncRNAs of group-1 lncRNAs such as AC019117.2 and LINC00941 was ascertained through GO and KEGG pathway analysis. As per the GO enrichment analysis, the target mRNA of LINC00941 mainly involves in the cell junction, cell adhesion, extracellular matrix (ECM), and cell growth, and fosters tumor cell invasion, metastasis, and growth (Figure 7(a)). Furthermore, results revealed that the ECM–receptor interaction, focal adhesion, PI3K-Akt, mitogen-activated protein kinase (MAPK) signaling pathways, PD-1 checkpoint signaling, and PD-L1 expression in KEGG pathway analysis were significantly involved in facilitating metastasis and tumor cell differentiation of GC (Figure 7(b)). This result was broadly consistent with that of lncRNA and mRNA interaction networks and previous studies. 43 , 45 Specifically, AC019117.2 in group-1 lncRNAs and its target mRNAs were conducive to the development of familiar biological features for tumor cell proliferation, as depicted in Figure 8(a) and (b), which demonstrated the validity of our study.
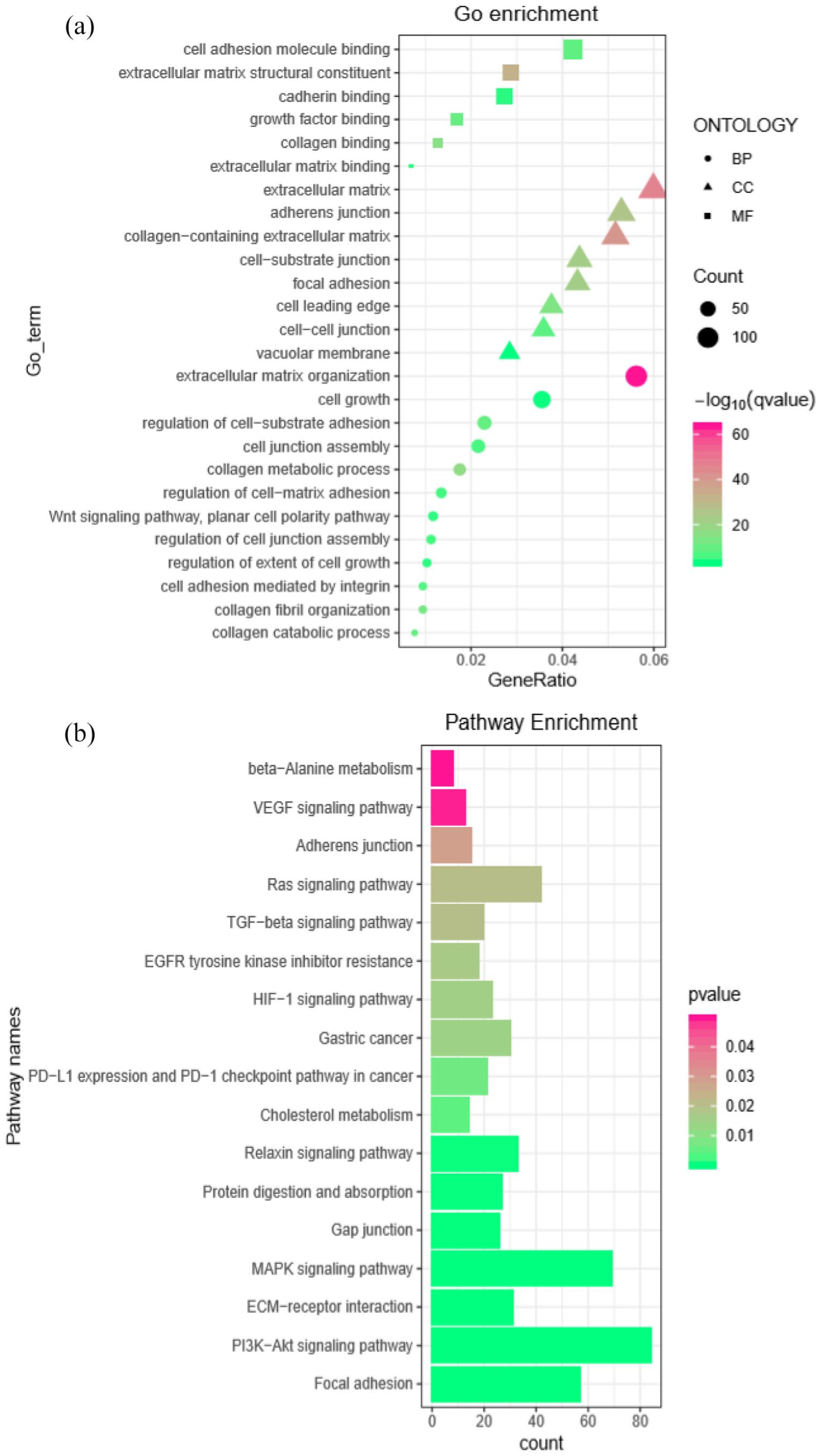
GO and KEGG pathway analysis for LINC00941. (a) The bubble plot showed a significantly enriched GO pathway. (b) The bar plot showed a significantly enriched KEGG pathway. LINC00941 and its target mRNAs were involved in modulating several signaling pathways to enhance GC progression. (A color version of this figure is available in the online journal.)
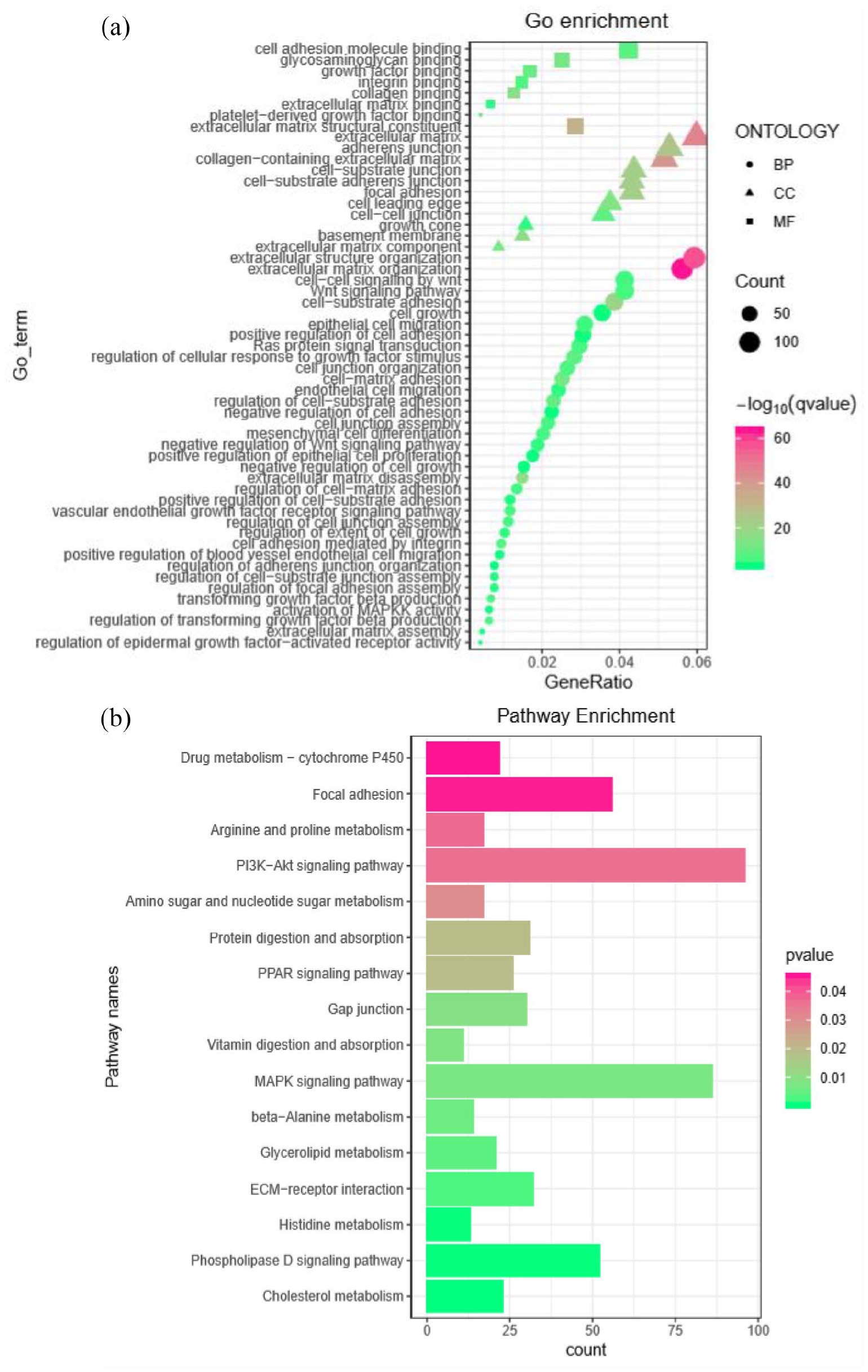
GO and KEGG pathway analysis for AC019117.2. (a) The bubble plot indicated a significantly enriched GO pathway. (b) The bar plot delineated a significantly enriched KEGG pathway. AC019117.2 and its target mRNAs were conducive to the modulation of several signaling pathways to enhance GC progression. (A color version of this figure is available in the online journal.)
Discussion
The lack of effective prognostic biomarkers to diagnose GC on the basis of clinical stage-wise still remains an urgent issue to explore significant modulators in the development of GC. 49 Over the past several years, several reports delineated that the expression of lncRNAs plays an essential clinical role in GC diagnosis. Different lncRNAs may play crucial roles in multiple genetic and epigenetic molecular networks in several cancers including GC as they are involved directly or indirectly in affecting cellular homeostasis, cell proliferation, migration, and stemness. 50 It is well known that the cancer pathological grade is an essential factor for prognosis. Higher level pathological grades unveil the faster growth, invasion, metastasis, and worse prognosis, and may require more timely aggressive treatment. It is possible to develop a panel of lncRNAs with variable expression as prognostic/diagnostic markers conducive to the tumor microregulatory networks of GC based on pathological grade or clinical stages. These lncRNAs are involved in the molecular signal transduction, and regulate specific genes and corresponding molecular signaling pathways pertinent to GC progression. Past studies focused to delineate the role of individual lncRNA expression within the primary tumor tissues and normal tissues; 51 yet, there are no studies reported for the effective screening of clinical stage-wise prognostic biomarkers and exploring their potential mechanisms associated with the exact clinical stage for GC patients. Therefore, the current study involves the analysis of lncRNA expression for developing clinical stage-wise prognostic markers pertinent to GC using TCGA and GEO databases.
Zhang et al. 52 demonstrated that abnormal histone modification–activated HOXC cluster antisense RNA 3 (HOXC-AS3) expression can foster the GC cell proliferation through the Y-box binding protein 1 (YBX1). Zhu et al. 9 presented a 24-lncRNA signature for the clinical prognosis of GC that might participate in cancer recurrence and metastasis. UCA1, MALAT1, HOXA11-AS, and ZEB1-AS1 are reported lncRNAs, which can modulate the progression of GC. Furthermore, a few studies reported the multifunctional roles of lncRNAs including FEZF1-AS1, HOTAIR, HOXA11-AS, HOTTIP, and LINC01234 involved in the progression of GC.29,53–56
Upregulation of lncRNAs such as CASC19, 57 SNHG6, 58 MIR100HG, 59 Sox2ot, 60 MALAT1, 61 NEAT1, 62 SNHG8, 63 and CTD-2510F5.4 directly or indirectly correlated to the clinicopathological parameters on TNM stage-wise GC progression. 50 For instance, the lncRNA growth arrest associated lncRNA 1 (GASL1) 64 and papillary thyroid carcinoma susceptibility candidate 3 (PTCSC3), and MALAT1 expression predominantly exhibited correlation with TNM stage, tumor size, and distant metastasis of GC, respectively.64–66 LncRNAs including “SNHG6, ARHGAP27P1 (Rho GTPase activating protein 27 pseudogene 1), DANCR (differentiation antagonizing non-protein coding RNA), DGCR5 (DiGeorge syndrome critical region gene 5), MT1JP (metallothionein 1 J, pseudogene), SNHG17, and ZFAS1” are significantly correlated with the TNM stage as well as tumor invasion depth, and lymph node metastasis pertinent to GC progression.67–73 HOXA11-AS and tubulin alpha-4b are the other significant lncRNAs expressed in GC, and their expression is correlated to TNM stage as well as lymph node metastasis of GC.74,75
AK026189 (an lncRNA) is highly expressed in GC; the expression of this lncRNA is significantly correlated to the reduction in OS. 9 H04858 is another lncRNA involved in regulating the oncogenes as well as hematopoiesis pertinent to myeloid leukemia progression; hence, the expression of H04858 could be considered as the early prognostic marker for leukemia. 76 Expression of AK026189 (CASC 15) is significantly associated with neuroblastoma and extensively enhanced in melanoma progression.77–79 Ren et al. 22 delineated that a total of 76 lncRNA GC-specific gene signatures were observed from pan-cancer analysis. Among them, total five lncRNAs such as “CTD-2616J11.14, RP1-90G24.10, RP11-150O12.3, RP11-1149O23.2, and MLK7-AS1” are predominantly involved in modulating the OS of GC patients as they can effectively regulate DNA replication, cancer cell mitosis, apoptosis, and RNA splicing. 79 In our study, the analysis of lncRNA expression profiles from TCGA database revealed that the total 529 upregulated and 107 downregulated lncRNAs were identified, and the expression profiles of lncRNAs expressed in tumor tissues as well as in the normal tissues were compared in order to delineate the significant correlation between lncRNAs and clinical stage-wise GC; group-1 lncRNAs including AC019117.2 and LINC00941 were downregulated in normal tissues and exhibited a positive correlation with the clinical stages of stage I to stage IV in GC; group-2 lncRNAs such as AC012317.2 and AC141273.1 were upregulated in normal tissues and reported typically a negative correlation with the clinical stages of stage I to stage IV in GC. Overall, LINC02410 expression was declined in different clinical stages of GC tumor samples than normal samples.
Minimal expression of HOXA11-AS in GC patients exhibited a good OS rate, whereas the minimal expression of tubulin alpha-4b exhibited a minimal OS rate.74,75 Furthermore, the expression of LINC00086 is correlated to a low OS rate as it could promote lymph node metastasis and TNM stage. 80 Kaplan–Meier analysis for these five lncRNAs showed that the low AC019117.2 and LINC00941 expressions can effectively predict better prognosis and could be considered as prognostic markers in order to predict the GC progression.
Substantial expression of AA041523 can induce loss of tumor suppressor LKB1 gene function, a specific mutational event occurring in lung cancer. 81 Furthermore, the expression of AA041523 is also predominantly higher in GC and fosters tumorigenesis, and the expression of this lncRNA is correlated with OS of the patient and tumor size. 82 As per our current study, LINC00941 among the five lncRNAs has been found to be considered as a prognostic biomarker due to its involvement in the GC development, invasion, and metastasis. 45 LINC00941 gene silencing can induce mitigation in the cell proliferation and migration of GC in vitro. 43 In addition to GC, LINC00941 also can regulate the incidence and development of head and neck squamous cell carcinoma, 83 lung adenocarcinoma,48,84 and hepatocarcinogenesis. 85 Our study reported that LINC00941 expression is positively correlated with tumor clinical stages I to IV, and the higher expression of LINC00941 exhibit has a significant relationship with poor prognosis. GO and KEGG pathway analysis for LINC00941 and AC019117.2 revealed that their target mRNAs are significantly involved in modulating the signaling mechanisms pertinent to tumor cell invasion, metastasis, and growth. LINC00941 was mainly enriched in many signaling pathways related to tumor cell invasion, metastasis, and growth. The lncRNA and mRNA interaction analysis depicted that the AC019117.2 expression was linked to LINC00941 through ITGA3 and TPBG, and it has been confirmed that TPBG can modulate cell adhesion and further associated with poor clinical outcomes for lung cancer patients. 86 Another study by Koshizuka et al. 87 found that knockout studies of ITGA3 in esophageal squamous cell carcinoma (ESCC) cells can prevent tumor cell migration and invasion; overexpression of ITGA3 has predicted poor prognosis for ESCC patients. However, its role in cancer development has not been reported. AC019117.2 might have a synergistic effect on LINC00941 via ITGA3 and TPBG to exert its role in the tumor cell biological process based on our results.
Overall, the systematic analysis of transcriptomic data in TCGA and GEO databases pertinent to GC models depicted the functional aspects of five lncRNAs signature in the GC tumor progression. This novel lncRNA expression based on the clinical stages of GC may be used to distinguish the tumor malignancy in GC patients and further provides a possible way to the early prognosis and early treatment of GC.
Footnotes
Acknowledgements
The authors convey their sincere thanks to later Prof. Gumrackh Aliev, Gally International Biomedical Research and Consulting LLC, San Antonio, Texas, USA, for the support during the entire work.
Authors’ Contributions
NMB, RF, JL, and VNN conceptualized and designed the study. NMB executed data analysis. NMB, HG, NX, YL, HY, and KC performed the literature analysis and wrote the original manuscript draft. NMB, JL, VNN, and RF performed editing and revision of the final draft. Every author reviewed and approved the manuscript draft prior to submission.
Declaration Of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by National Natural Science Foundation of China (No. 81703158), and Russian Academic Excellence project “5-100” for the Sechenov University, Moscow, Russia, GALLY International Research Institute, San Antonio, Texas, USA.
Data Availability
Data obtained for this study will be provided upon request by the journal office and reviewers.