
Abstract
DNA barcoding is a method to identify biological entities, including individual cells, tissues, organs, or species, by unique DNA sequences. With the advent of next generation sequencing (NGS), there has been an exponential increase in data acquisition pertaining to medical diagnosis, genetics, toxicology, ecology, cancer, and developmental biology. While barcoding first gained wide access in identifying species, signature tagged mutagenesis has been useful in elucidating gene function, particularly in microbes. With the advent of CRISPR/CAS9, methodology to profile eukaryotic genes has made a broad impact in toxicology and cancer biology. Designed homing guide RNAs (hgRNAs) that self-target DNA sequences facilitate cell lineage barcoding by introducing stochastic mutations within cell identifiers. While each of these applications has their limitations, the potential of sequence barcoding has yet to be realized. This review will focus on signature-tagged mutagenesis and briefly discuss the history of barcoding, experimental problems, novel detection methods, and future directions.
Impact Statement
The work that we are submitting is important to the field by highlighting the contributions of DNA barcoding technology in taxonomy, functional toxicology, cancer biology, and cell lineages. The work advances the field by showing how conventional methods in barcoding can be developed or adapted to expand horizons in functional toxicology and cancer therapeutics. While this article is a minireview, it highlights latest information that has been obtained by the advanced technology. This latest information includes how profiling the yeast genome using novel “humanized” libraries has revealed information concerning the function of DNA damage tolerance genes in conferring toxicant resistance.
Introduction
DNA barcodes serve as biological identifiers much as 11-digit product codes serve as identifiers at the retail market. The DNA sequence that constitutes a barcode can derive from an internal sequence within a gene or be designed and synthesized in vitro and inserted into predetermined chromosomal locations in vivo. Considering that the four existing nucleotides can be arranged in any order to design a barcode, there are theoretically 4n potential barcodes, where n is the number of bases. Thus, 15 base pairs could yield over billion codes, which are enough to identify all planetary species.1 –3 Established technology to amplify barcode sequences and quantify relative numbers of barcode sequences by deep sequencing has enabled investigators to profile genomes and trace lineages after exposure to myriad stress conditions. Barcode technology has rapidly accelerated progress of biological queries. In this review, we discuss barcode applications in identifying organisms, cells, and genes. While the technology has made impressive advances, each application presents limitations. Current efforts to overcome these limitations include novel techniques to increase barcode diversity, bioinformatic tools and pipelines to minimize false discoveries, and novel sequence platforms to accelerate data acquisition. In this review, we discuss barcode applications in taxonomy and species identification, signature-tagged mutagenesis, cancer lineage studies, in-cell developmental studies, and viral genome sequencing (Table 1). Finally, we discuss novel applications and future directions. The following links give a general overview of barcoding, including applications and methods (https://www.youtube.com/watch?v=wKt0sAV51Xs; https://www.youtube.com/watch?v=bMgkMroXD5U).
Barcode applications and limitations.
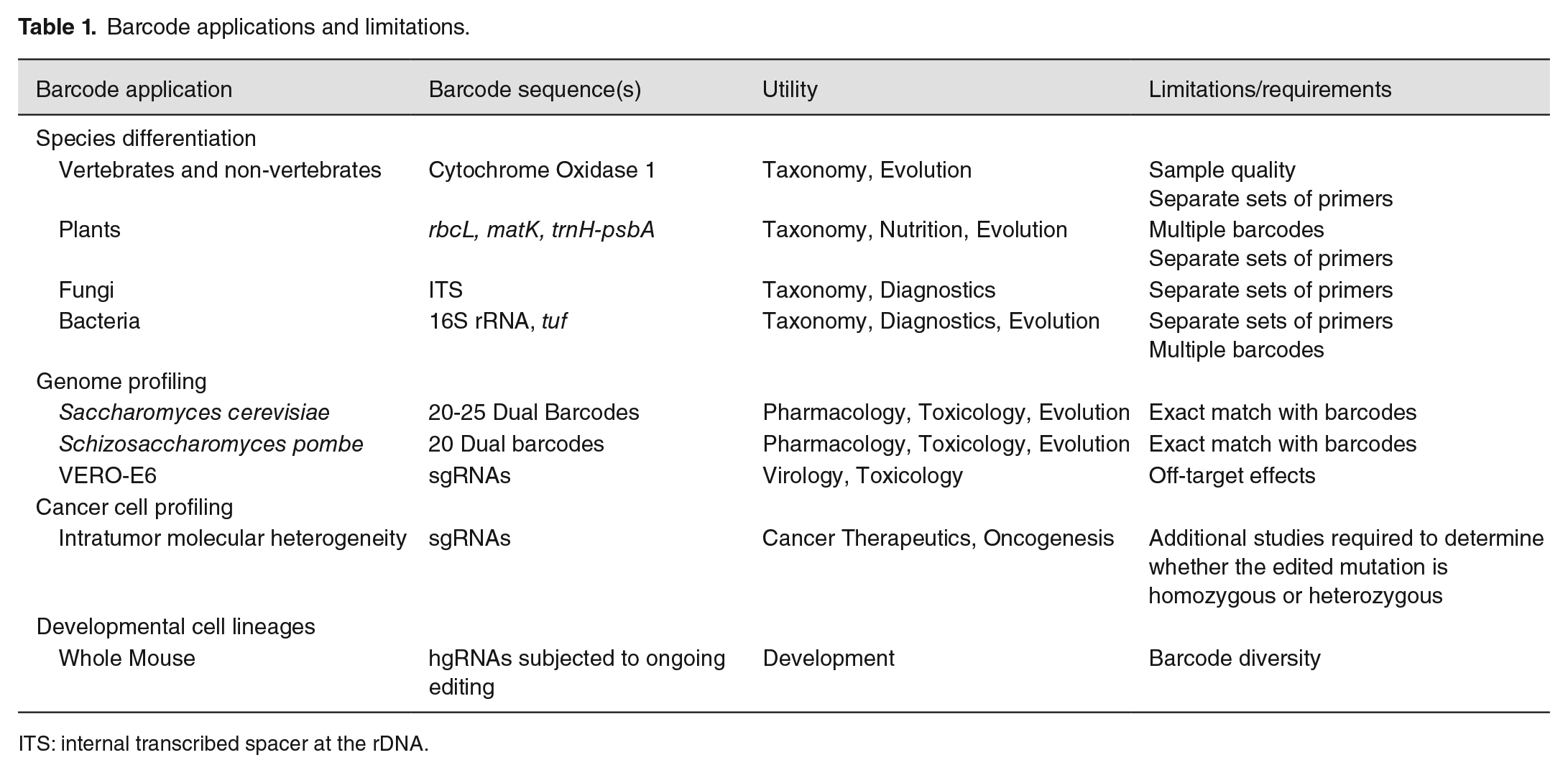
ITS: internal transcribed spacer at the rDNA.
Species identification
Hebert et al. 4 first envisioned DNA barcoding for distinguishing species. They reasoned that identifying over 10 million species by conventional taxonomy would expend an exorbitant amount of time and labor; however, microgenic characterization could be possible using available tools of polymerase chain reaction (PCR) and DNA sequences. For microgenic barcoding to succeed, the DNA sequence differences between species must be more than that within a species. 4 Hebert et al. 4 used the 650 bp from the 5′ end mitochondrial cytochrome C oxidase 1 (CO1) as a signature sequence that can distinguish 200 closely allied species of lepidopterans and other specimens. The technique rapidly spread to the barcoding of other vertebrate and non-vertebrate species, resulting in approximately 4000 published papers from 2003 to 2019. 5 The basic technique utilizes standard methods in molecular biology, including DNA extraction, PCR amplification, and DNA sequencing. DNA sequences are then compared using GenBank data sets or barcode of life data set (BOLD).6,7
While CO1 barcoding has provided species-level specificity for mammals, fish, and birds, retrieving full-length barcodes may be difficult. 8 First, DNA may be degraded from stored samples or processed food. Second, distinct primer sets are required. To economize effort, other approaches have shown that shorter regions of CO1 (200 bp) are sufficient. 8 These mini-barcodes are effective in archival specimens and have been used for characterizing food products and pharmaceuticals.9,10
Taxonomic barcoding for bacteria, plants, and fungi requires different sequences. For plants, these have included ribulose biphosphate carboxylase (rbcL), maturase kinase (matK), transfer RNA-H and photosystem II D1-arabidopsis thaliana (trnH-psbA), and internal transcribed spacer at the rDNA (ITS).11,12 For bacterial strains, 16S ribosomal RNA (16S rRNA), elongation factor Tu (Tuf gene), and chaperonin have been used as signature sequences; many studies have relied on nine hypervariable regions (V1-V9) in the 16S rRNA sequence.12 –14 For fungi, nuclear ITS at the rDNA has been used as a signature sequence. 13 These barcodes are now complementing studies performed in taxonomic research, population genetics, and phylogenetics. 15 Barcoding technologies have been especially beneficial in enhancing the speed and quality of diagnostics and identifying novel species, where normal identification of bacterial pathogens depends on culturing and phenotyping clinical isolates.
While 16S rRNA barcoding is extensively used in identifying microorganisms, it has limitations in quantifying and identifying species within the microbiome. The 16S rRNA genes are variably repeated in bacterial species, and thus, species with low copy number may be undercounted.16,17 In addition, hypervariable 16S rRNA sequences may be difficult to amplify by PCR due to high GC content, rendering a bias in reading the 16S rRNA regions.16,17 Currently, 90% and 86% of bacterial species can be recognized at the genus level and species level, respectively.18,19 Genus recognition can be enhanced by complete 16S rRNA sequencing using platforms, such as Oxford Nanopore Technologies (ONT) MinION.18,19
Signature-tagged mutagenesis
Signature-tagged mutagenesis studies20,21 have become crucial in understanding the functions of genes uncovered by DNA sequencing. In the last 25 years alone, 3278 unique animal species across 24 phyla have been sequenced.3,22 However, the sequence data alone often fail to provide insights into the phenotypes of many open reading frames (ORFs). Even in a well-established model organism, such as Escherichia coli, many ORF functions have yet to be defined. 23 The search for mutant phenotypes to discern gene function is laborious if each ORF is individually studied. An alternative strategy is to pool strains in which individual mutants are identified by a synthetic molecular barcode in a unique ORF. These synthetic barcodes can be positioned using signature-tagged mutagenesis (STM). Pertinent to the focus of this review is the use of barcoding to identify drug targets and xenobiotic resistance.
STM has been used for multiple microorganisms, including E. coli, 24 Saccharomyces cerevisiae (budding yeast),25,26 Schizosaccharomyces pombe (fission yeast),27,28 Candida albicans, 29 and Candida glabrata. 30 Techniques to mutagenize these strains include in vivo transposition (E. coli), 24 gene replacement by homologous recombination (S. pombe, 28 budding yeast), 25 and non-homologous (illegitimate) recombination (S. pombe, 27 C. glabrata). 30 The limitations in tagging every ORF include zygosity, essential genes, and gene duplications. Stable haploid S. pombe and budding yeast strains have rendered it possible to construct both haploid and diploid deletion libraries of strains contain knockouts of non-essential genes. 23 While essential genes cannot be completely knocked out, knockdown alleles can be made in budding yeast using the decreased abundance by mRNA perturbation (DAmP) approach. 31 In addition, haploid insufficiency libraries are available that consist of diploid strains that are heterozygous for known knockdowns. In total, the yeast deletion collections contain over 21,000 mutant strains. 32
These pooled collections can then be screened for genes that confer resistance or sensitivity to antibiotics, pharmaceuticals, toxicants, nutrients, temperature (heat or cold shock), hypoxia, oxidative species, and other environmental conditions.33 –36 The advantage of using the model eukaryotes S. pombe and budding yeast are that many genes are orthologous to higher eukaryotic cells; indeed Kachroo et al. 37 demonstrated that many essential genes could be replaced by the corresponding human gene and restore function. These screens are even more important now considering that 42 billion pounds of chemicals are produced or imported into the United States of America daily. 38
The design of the barcode cassette is typically a conserved sequence (drug resistance) flanked by 20-25 bp unique sequences (Figure 1). 25 In budding yeast, these unique sequences are referred to as uptag and downtag sequences. 39 These can then be amplified by PCR and sequences counted by high throughput sequencing platforms, including Illumina HiSeq and MiSeq platforms. 40 Typically, PCR reads include indices that allow for highly multiplex pooled experiments. 41 Relative numbers of barcodes obtained from cells with and without treatments can then be processed by various bioinformatics pipelines to identify genes that confer resistance or sensitivity to various DNA damaging agents.
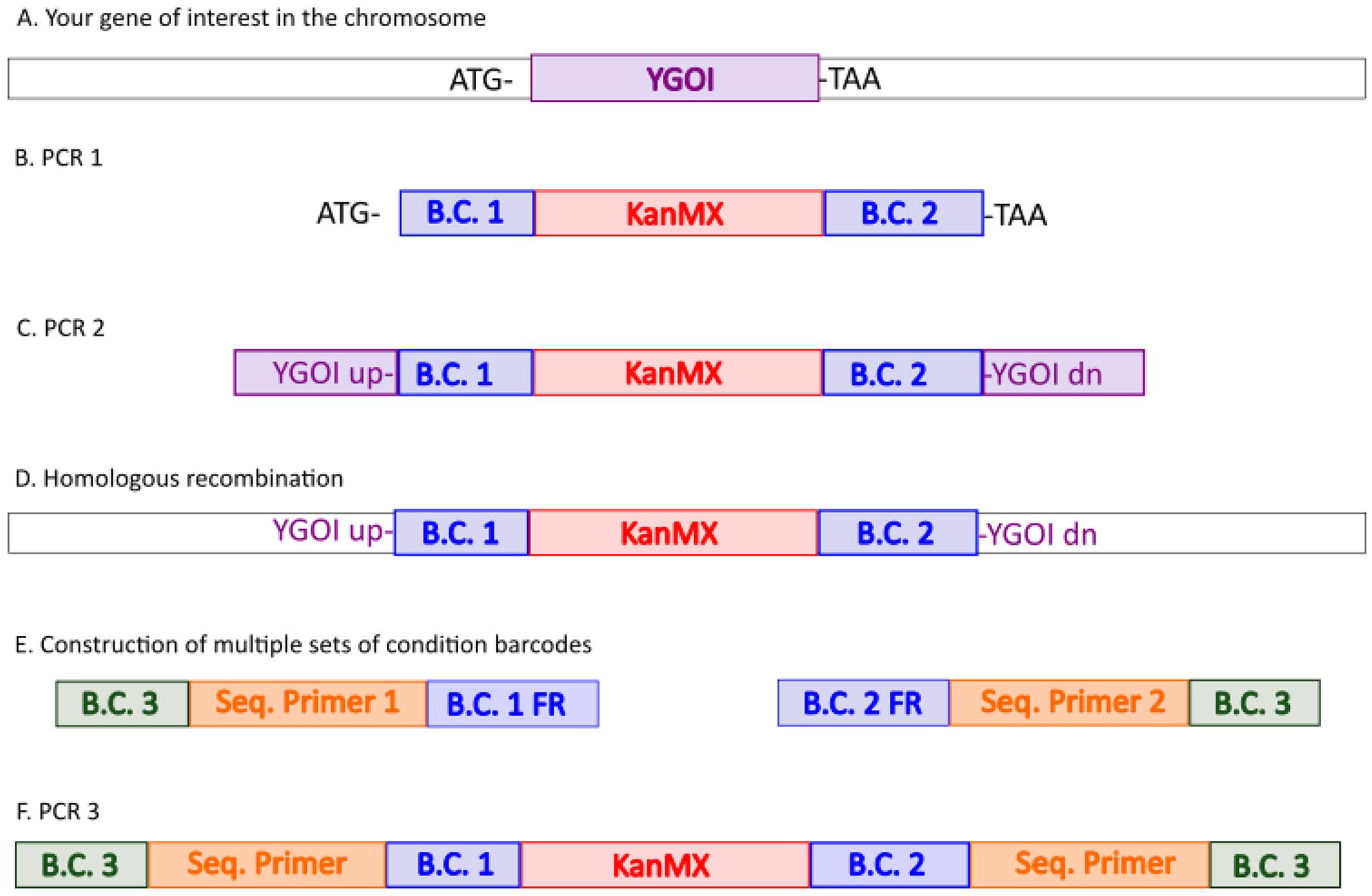
Barcoding strategy for multiplexed high-throughput screens in yeast. The chromosome context your gene of interest (YGOI) is unfilled, and other sequences are colored blue, red, orange, and purple. (A) Open reading frame (ORF) of YGOI is bordered by the initiation codon (ATG) and termination codon (TAA). (B) Unique Uptag (BC 1) and downtag (BC2) are added by PCR and flank the KanMX cassette. (C) A second PCR reaction (PCR 2) includes homologous sequences both upstream and downstream of YGOI. (D) The PCR 2 product is used to knock out YGO1 by homologous recombination and selection for KanR isolates. (E) Construction of oligomers containing the sequencing primer sequence (Seq. Primer 1 and 2), flanked by experimental barcode (BC 3) on one side to denote treatment condition, and BC 1 or BC 2 flanking regions (FR) on the other side. (F) In PCR3, oligomers from E are used to generate PCR fragments that are then sequenced using the applicable sequence platform. The figure is an adaptation from Giaever et al. 33 (A color version of this figure is available in the online journal.)
A typical processing pipeline goes through several steps: quality assessment, demultiplexing (if necessary), read trimming, quality filtering, barcode counting, and barcode count preparation and analysis. Multiplexing allows for multiple conditions to be tested individually and sequenced in one run, decreasing time and cost. Read trimming and quality filtering accelerate the barcode counting processes by reducing the amount of data to run through the program. The counting program compares sequencing reads to a list of barcodes and matches them with their corresponding ORFs. A tolerance of 0 or 1 mismatches is useful to minimize false positives. 42 The counts are then prepared by aligning counts for each barcode from each replicate of a control and a treatment, followed by normalization and analysis using preferred programs. 43 One way to do this is to treat the analysis as an RNASeq pipeline. An example process flow would include importing the data into R and using the TCC package 44 to normalize data with a trimmed mean of m-values (TMM), followed by determining differential expression by negative binomial regression analysis with edgeR,45 –47 allowing for a floor PDEG of 0.05 and FDR < 0.1, and iterating three times.
One limitation of the library for drug analysis is that budding yeast does not have the metabolic capacity to activate many chemicals. 48 This is particularly important since many prodrugs and xenobiotics require cytochrome P450 enzymes to bioactivate compounds. In the case of toxins, these bioactivated compounds include reactive epoxide derivatives. One way to circumvent the bioactivation requirement is to use the fully activated compound. 49 The disadvantage to this approach is that the activated compound may interact with external cellular constituents. Another way is to “humanize” the collection by introducing human cytochrome P450 (CYP) genes into the deletion collection. Yeast vectors are available that over-express CYP1A2, 50 CYP3A4, 51 CYP1B1, 52 and CYP1A1. 50 These expression vectors can be introduced by conventional lithium acetate-mediated DNA transformation. 53 CYP-containing transformants also exhibit robust CYP activities. An alternative approach for introducing genes into pooled strains is selective ploidy ablation (SPA). 54 As an example, St John et al. 55 introduced CYP1A2 into the non-essential yeast deletion collection. They then profiled the humanized yeast deletion and the original yeast deletion collection for aflatoxin B1 (AFB1) resistance. While only one gene from the original yeast deletion pool lacking CYP1A2 was identified as conferring resistance to AFB1, 86 genes were identified in the yeast deletion collection expressing CYP1A2. 55
Currently, only a few of the 57 human CYP genes have been expressed in budding yeast. 48 While the human CYP1 family functions to bioactivate large molecular weight (MW) aromatic compounds, other CYPs, such as CYP2E1, function to activate small MW compounds, such as ethanol and acetaminophen. 56 In addition, other xenobiotics require activation by multiple human enzymes; examples include polyaromatic compounds that require epoxide hydrolase and CYP1A1, 57 and heterocyclic aromatic amines that require CYP1A2 and n-acetyltransferases (NAT2). 58 A potential limitation of this technique is that expression of the P450s in yeast is confers a slower growth phenotype, possibly due to oxidative stress. 59 Thus, the future challenge is to co-express multiple CYPs and human enzymes without compromising growth.
While budding yeast has been used extensively in functional profiling studies, S. pombe also contains genes orthologous to human genes, such as NEIL1, which are not present in budding yeast. 60 A S. pombe deletion library has been constructed.27,28 And both homozygous and heterozygous deletion collections are available. 29 These collections have been shown to be useful for identifying drug targets. For example, the anticancer drug sunitinib, a tyrosine kinase inhibitor, has a strong cardiotoxicity side effect. Screening a heterozygous deletion collection for sensitivity to sunitinib identified the mitochondrial DNA polymerase (POG1) as a target. 61 A corresponding knockout of the POLG gene in human cell lines also conferred cytotoxicity, suggesting that the mitochondria was a key target in conferring the strong side effects. 61
Libraries for screening for drug targets in pathogenic fungi and bacteria
The yeast C. albicans is pathogenic in humans, and antifungal drug resistance is an emerging problem, especially among immunocompromised patients. Determining drug targets, as performed using pooled libraries of budding yeast, is complicated since diploid but not haploid strains can be cultured in the laboratory. Since the budding yeast and C. albicans genomes are similar, 62 one approach is to perform screens in budding yeast and then knock-out the corresponding gene in C. albicans. For example, the budding yeast TORC1 pathway is required for sensitivity to the ergosterol-targeting fungicide amphotericin B (AmB) in both biofilm and planktonic cells. 34 Screening for resistance to AmB revealed that the two growth modes had significant overlap in AmB-persistent mutants, including mutants defective in sterol metabolism, ribosome biosynthesis, and TORC1. In C. albicans and C. glabrata, rapamycin-mediated inhibition of TORC1 also increased AmB resistance. 63 Thus, budding yeast drug targets provide insights into possible targets in pathogenic Candida strains. 63
A secondary approach is to use a partial knock-out collection that is available in C. albicans and score for haploid insufficiency. In this approach, Xu et al., 64 used ~2700 heterozygous deletion mutants for profiling the genome for fluconazole, voriconazole, caspofungin, 5-fluorocytosine, and AmB resistance. They identified targets in ergosterol, fatty acid and sphingolipid biosynthesis, microtubules, actin, secretion, rRNA processing, translation, glycosylation, and protein folding. This approach thus complements other studies to profile drug targets.
The same strategy to profile fungal genomes for antifungal resistance can also be used for to screen for antibiotic resistance in bacterial strains where knockout collections are available. The advantage of E. coli is that it is a well-established bacterial model and deletion collections are available.65,66 Liu et al. 66 profiled the bacterial genome for resistance to 22 antibiotics, including spectinomycin, cephradine, aztreonam, colistin, neomycin, enoxacin, tobramycin, and cefotoxin. 65 They identified 283 resistant strains which could be grouped into strains that exhibit multiple drug resistances and those that exhibit resistance to single drugs or drugs of the same category. 66
Use of CRISPR/CAS9 in constructing mammalian cell libraries for toxicogenomics
While the yeast and other fungal deletion collections are powerful tools for toxicogenomics, there is no equivalent deletion library for higher eukaryotic cells. An approach has been devised to use clustered regularly interspaced short palindromic repeat (CRISPR) technology that use single guide RNAs (sgRNAs) to knockdown (KO) mammalian genes.67,68 These sgRNAs can thus serve a dual purpose of interfering with a cell-specific function and introducing a unique sequence barcode, which can be identified by short-read sequencing.69 –71
Useful higher eukaryotic cell lines for developing these screens include Chlorocebus aethiops sabaeus (green monkey) kidney epithelial cell line (VERO-E6). For example, the genome of the C. a. sabaeus has been determined and VERO-E6 sequenced. 72 These resources were used to design genome-wide libraries to knockdown ~19,053 genes identified in this species.72,73 In brief, Grodzki et al. 73 used a domain-targeted CRISPR KO approach in which sgRNAs are identified that target active sites or functional domains of each gene. 74 About four sgRNAs for each of the ~19,053 genes were synthesized. In total, ~76,212 sgRNAs and 500 non-targeting control sgRNAs (Custom Array) were cloned into the LentiCRISPRv2 Puro (Addgene) vector to generate the AGM CRISPR KO library. sgRNA oligos are amplified from the plasmid pool by PCR and deep sequenced by next generation sequencing (NGS) to confirm adequate sgRNA representation. The library, also referred to as the Brunello library, 75 is packaged into lentivirus by standard methods. This library is then transfected into VERO-E6 cells and a stepwise puromycin selection is performed to establish stable transfectants. The transfectants are then exposed to the toxicant or the vehicle alone for defined time periods, generally seven to fourteen days.
Like data acquisition for yeast barcode experiments, read quality checks are performed with FASTQC tools. To align the processed reads to a library of interest, the designed sgRNA sequences from the library are compressed by transforming them into a Burrows-Wheeler index using the build-index function in Bowtie. 76 The Burrows–Wheeler index is a compressed version of the original sequence data that allows for accelerated alignment of short reads to a reference genome. 76 After alignment, alignment efficiencies are checked and the number of uniquely aligned reads for each library sequence is calculated creating a table of raw counts. Using the edgeR 45 and limma voom R packages, 77 the count table is input for analysis. The raw counts are normalized with the upper-quartile normalization method in which the scale factors are calculated from the 75% quantile of the counts for each sample, after removing genes which are zero in all samples. 44 Differentially expressed genes are identified, using the negative binomial approach implemented in edgeR, the corresponding continuous approximation in limma voom 77 or the CRISPR analysis software MAGeCK. 73 Finally, a consolidated annotated summary table is created showing all candidate sgRNAs differentially represented at FDR < 0.05. One problem with using CRISPR/CAS9 for gene targeting is off-target effects. This is mitigated by using multiple sgRNAs. Additional validation can also be performed by measuring toxicant resistance after silencing a specific gene using a short hairpin RNA (shRNA).
Several successful genome-wide CRISPR screens have been performed to identify resistance to SARS-CoV-2 (coronavirus) in VERO-E6 cells and in HEK293 cells. 73 These studies could be expanded for profiling genomes for resistance to other genotoxins. For example, studies have been performed to profile the human hematopoietic K562 cell line for formaldehyde resistance. 78 Comparison with similar studies in yeast have revealed shared resistance genes. 79 Ongoing studies are expanding these studies to additional toxicants. However, one caveat is that it is unknown whether VERO-E6 cells and HEK293 cells are equally capable of bioactivation of prodrugs and toxicants. Further studies are thus needed in profiling the metabolic activities of these cell lines. 45
Cancer cell barcoding
While the signature-based mutagenesis schemes in VERO-E6 cells and microorganisms are valuable in studying drug targets and identifying genes that confer resistance and sensitivity to toxins, signature-based mutagenesis in cancer cells is aiding in the identification of driver mutations that confer resistance to chemotherapeutic drugs by promoting proliferation and metastasis. Cancer cells exhibit accelerated genetic instability, and it is unclear whether resulting mutations can drive growth or are merely spectators. 80 These strategies are now being implemented to guide clinicians in treatment of advanced stage pancreatic cancer 81 and head and neck cancers. 82
The basic scheme to address the question is to use CRISPR/CAS9 to introduce both silent mutations and known mutations into the cancer cells and then trace which cells become more proliferative. This not only identifies the selected mutations but also the mutations that drop out and are therefore not important for the increased proliferation.
As an example, Guernet et al. 80 devised modified cancer cell lines using CRISPR-Barcoding to investigate the resistance to epidermal growth factor receptor (EGFR) inhibitors. To test their model, they used an EGFR-T790M mutation that confers resistance to gefitinib, an ATP-competitive EGFR inhibitor and an EGFR-T790T mutation, which served as a silent mutation. EGFR-T790M expressing cells were detected by quantitative PCR. They showed that EGFR inhibitor treatment of a mixture of these cells preferentially enriched for EGFR-T790M, as expected. Considering that mutations in multiple genes could also confer EGFR resistance, the investigators also devised multiplex models for EGFR inhibitor resistance, including RAS mutations, EML4-ALK fusions, and EGFR T790M; all these cell lines were enriched. The results obtained in cell culture were also supported by results obtained when cell mixtures were implanted in nude mice and the relative proportion of cell genotypes were determined from the resulting tumor. The disadvantage of the system is that it was unknown whether the CRISPR-generated cell lines were heterozygotes or homozygotes for each mutated gene. 80
One interesting extension of these studies is that mutations in known oncogenic drivers can also be introduced in drug-resistant cell lines to determine whether these mutations would decrease resistance or proliferations in advanced cancer cells. For example, Guernet et al. 80 showed that knocking down the APC gene was sufficient to reduce growth of advanced cancer cells. 83 Thus, this technique has potential for identifying oncogenic drivers in advanced cancer, where drug resistance presents treatment challenges.
Whole organism barcoding to determine cell lineage
The advent of barcoding and CRISPR/CAS9 technologies with the versatility of high-throughput sequencing has also incited interest in studying cell lineages. While cell lineage studies have been performed for model organisms such as Danio rerio (zebrafish) 84 and Caenorhabditis elegans (C. elegans), 85 cell lineage studies for more advanced eukaryotic models have not been forthcoming. This presents a challenge in human cell lineage studies, considering that the adult human has 37 million cells, of multiple types, which are all descended from a single zygotic cell. 86 Gaj and Perez-Pinera 87 and Kalhor et al. 88 used a modification of the shRNA technique to create cell-lineage specific barcodes. They reasoned that the origin of cell lineages could be traced if the terminal lineages were marked by diverse barcoded sequences that “evolved” from common incipient barcode sequences, analogous to a tree emanating branches, stems, and leaves. To generate the “evolved” barcode sequences, they used a molecular tool that was modeled on a self-targeting CRISPR/CAS9 homing guide RNA (hgRNA). While similar to single guide RNAs, these hgRNA encode a protospacer adjacent motif (PAM) that enables Cas9 to target the expression of the cassette encoding the hgRNA. 88 Kalhor et al., 88 created a transgenic mouse, designated MARC1 (mouse for actively recording cells) that had 41 different hgRNA expression cassettes that were integrated in the genome. These expression cassettes were localized to introns or intergenic regions of the genome and did not interfere with normal mouse development. They then crossed this MARC1 mouse with a mouse that contained an expressed Cas9 transgene. The basic idea was that the Cas9 would self-target and introduce stochastic mutations by non-homologous end joining (NHEJ), referred to as in vivo barcoding. The technique could generate 1023 barcodes using ten different hgRNAs, sufficient to barcode approximately 1010 cells in the mouse. 88 By high-throughput sequencing, Kalhor et al. 88 identified cells of common lineages by identifying similar mutation profiles (see Figure 2). They were able to generate a lineage tree for the early developmental stages in four embryos. The limitation of this study is that only a narrow spectrum of non-random mutations was generated.
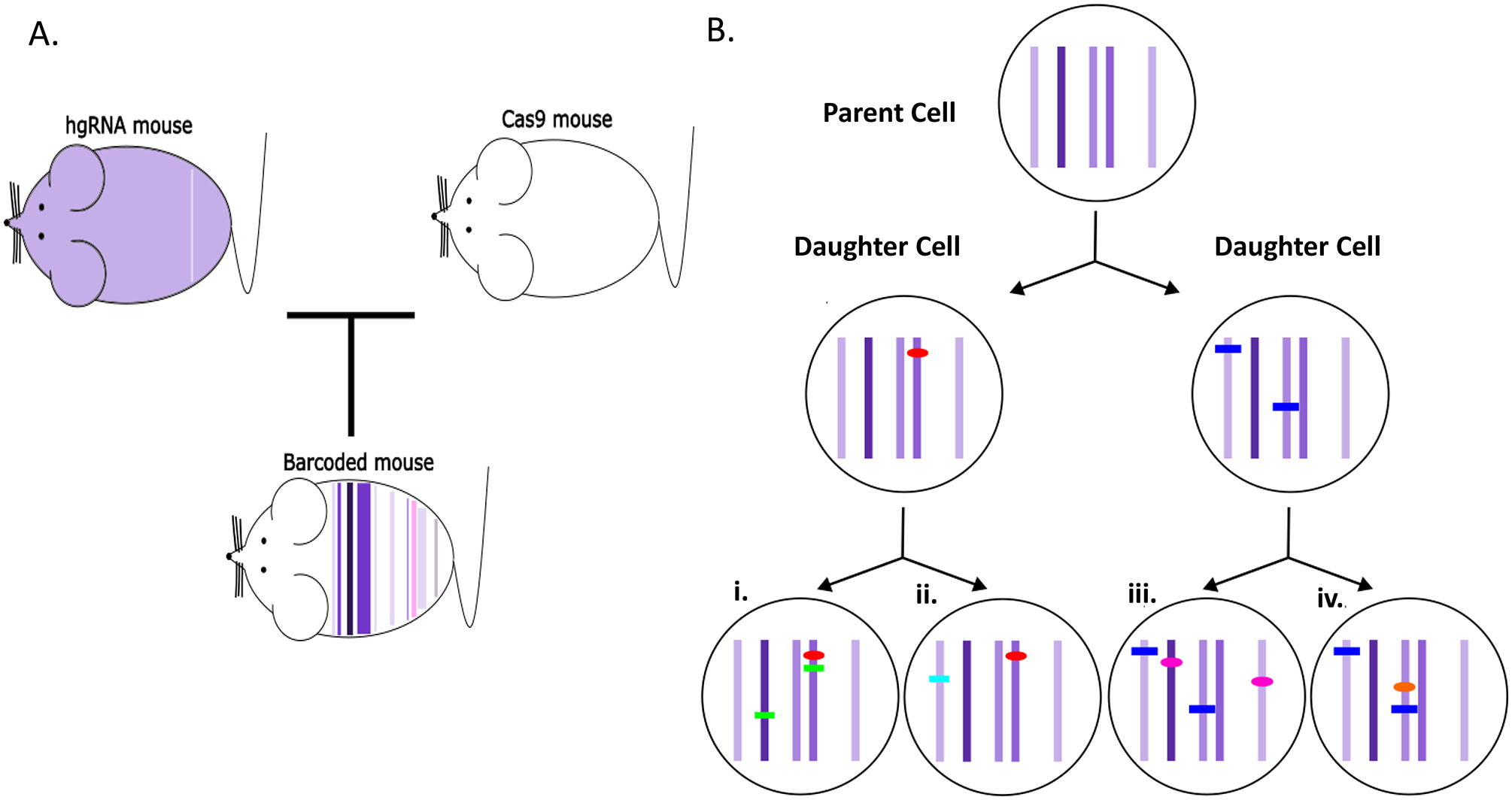
The methodology for generating a mouse where cell lineages are barcoded. In panel
To generate even more diverse barcodes to represent more sequences, Halperin et al. 89 converted Cas9 into a “nickase” that cleaves only one strand of the target DNA sequences fused to an error-prone and nick-translating E. coli DNA polymerase I. Similar to the previous study, 87 this Cas9-derivative could be targeted to a particular sequence of interest. In contrast, it generates edited sequences in 350 bp, which enables better randomization and reduces the number of target sites. EvolvR has also been used in bacterial cells, and a variation of the technique has also been used in budding yeast. 90 Thus, there are multiple potential applications in a variety of cells.
Mutation profiling of viral genomes
Severe acute respiratory syndrome coronavirus-2 (SARS CoV-2) is a relatively new pathogen that causes COVID-19 and is responsible for over a million deaths in the United States of America. 91 The SARS CoV-2 has rapidly evolved to evade the immune system and spread in the population. Thus, there is an urgent need to rapidly sequence variants to identify new emerging viral strains. One method involves sequencing 1.2 kb tiled amplicons using rapid barcoding kits.92,93 Two PCR reactions are performed for each patient sample; each reaction involves a set of primers that generate non-overlapping fragments. The two sets of PCR fragments form a tile array with minimum overlapping sequences, thus enhancing PCR efficiency.
Hypermutable regions of the viral genome can thus be identified by whole viral genome sequencing. Laha et al. 94 observed that frequent mutations appeared in the glycoprotein, nucleocapsid, ORF1ab, and ORF8. However, mutations in the envelop, membrane, ORF6, ORF7a, and ORF7b conserved the amino acid composition. In all, they identified 20 viral variants. 94 Additional studies are currently being pursued by epidemiologists. Identifying such mutations will aid clinicians and pharmacologists in designing drug therapies that target specific transmissible variants.
Future challenges and novel applications
Barcoding has had a significant impact on diverse fields, ranging from toxicology to viral genome characterization, cancer biology, microbiome, and developmental lineages. These applications are being accelerated by new sequencing platforms and modifications of existing libraries of barcoded cells. For example, advances in toxicogenomics are being accelerated by humanizing existing barcoded yeast strains by expressing CYP genes; the ability to express all 57 CYP genes in budding yeast, similar to that achieved in fission yeast, 95 will potentiate the toxicological characterization of novel xenobiotics and pharmaceuticals.
The technology has now opened horizons for mapping the fate of individual cells within complex organisms for identifying which mutations are driving cancer proliferation in the context of highly heterogeneous mixture of cells. Future studies will demonstrate whether barcodes can identify all the cells of a mammalian organism, rendering it possible to address questions concerning development, aging, and cancer. Such studies may advance personal medicine by identifying genotypes of drug resistant cancer cells. While barcoding has rendered it conceivable to identify every cell in an organism, it is also advancing studies in how physiology and the environment influence the microbiome. Such studies will be useful in understanding the impact of pharmaceuticals, climate change, and space travel on complex microbiomes impacting human health.
Footnotes
Authors’ Contributions
Both authors contributed to writing the main text.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health (R15ES023685-03).