
Abstract
Epidemiological evidence links lower air quality with increased incidence and severity of COVID-19; however, mechanistic data have yet to be published. We hypothesized air pollution-induced oxidative stress in the nasal epithelium increased viral replication and inflammation. Nasal epithelial cells (NECs), collected from healthy adults, were grown into a fully differentiated epithelium. NECs were infected with the ancestral strain of SARS-CoV-2. An oxidant combustion by-product found in air pollution, the environmentally persistent free radical (EPFR) DCB230, was used to mimic pollution exposure four hours prior to infection. Some wells were pretreated with antioxidant, astaxanthin, for 24 hours prior to EPFR-DCB230 exposure and/or SARS-CoV-2 infection. Outcomes included viral replication, epithelial integrity, surface receptor expression (ACE2, TMPRSS2), cytokine mRNA expression (TNF-α, IFN-β), intracellular signaling pathways, and oxidative defense enzymes. SARS-CoV-2 infection induced a mild phenotype in NECs, with some cell death, upregulation of the antiviral cytokine IFN-β, but had little effect on intracellular pathways or oxidative defense enzymes. Prior exposure to EPFR-DCB230 increased SARS-CoV-2 replication, upregulated TMPRSS2 expression, increased secretion of the proinflammatory cytokine TNF-α, inhibited expression of the mucus producing MUC5AC gene, upregulated expression of p21 (apoptosis pathway), PINK1 (mitophagy pathway), and reduced levels of antioxidant enzymes. Pretreatment with astaxanthin reduced SARS-CoV-2 replication, downregulated ACE2 expression, and prevented most, but not all EPFR-DCB230 effects. Our data suggest that oxidant damage to the respiratory epithelium may underly the link between poor air quality and increased COVID-19. The apparent protection by antioxidants warrants further research.
Keywords
Impact Statement
Our study examined the impact of combustion-related oxidant particles, known as environmentally persistent free radicals (EPFRs), on SARS-CoV-2 infection of well-differentiated nasal epithelium, grown from human primary nasal epithelial cells at the air-liquid interface. We showed that EPFR exposure prior to SARS-CoV-2 infection induced epithelial damage, increased expression of a surface receptor responsible for activating the SARS-CoV-2 spike protein, increased viral replication in epithelium, increased proinflammatory cytokine secretion, and dysregulated cellular antioxidant pathways. Most but not all these effects could be prevented by pretreatment with the antioxidant astaxanthin. Taken together, these data suggest that induction of oxidative stress in respiratory epithelium may underly the associations between poor air quality and an increased prevalence and severity of COVID-19. Astaxanthin is a readily available dietary antioxidant that warrants further study, including clinical trials, to determine whether it can mitigate the effects of air pollution on respiratory diseases, including viral infections.
Introduction
Since the early reports of an increase in COVID-19 mortality associated with worse air quality in the USA, 1 this topic has attracted much attention. Overall, there is strong epidemiological support for an association between worse air quality and increased case rates, increased severity, and increased case fatality rates with COVID-19 in both high-income countries and low- and middle-income countries.1 –5 There is also support for acute exposures to air pollution associated with increased case rates, suggesting increased viral transmission,2,3,5 whereas longer term exposure was more associated with increased case severity and fatality.3,4 Short-term exposure to wildfire smoke has been associated with an increased SARS-CoV-2 positive test rate in the exposed community, 6 and to an increase in both cases and deaths from COVID-19. 7 In addition, a reduction in COVID-19 deaths was reported when traffic-related air pollution decreased associated with “lock-downs” in the USA and India.8,9
The adverse impacts of poor air quality on respiratory health are well known; however, the precise mechanisms are not entirely clear. Certainly, there is evidence that children subjected to poor air quality show biomarkers of oxidative damage in exhaled breath. 10 In addition, the adverse effects of air pollution exposure are magnified in those with reduced or null function variants in genes related to antioxidant defense.11 –13 Taken together, these reports demonstrate that at least some of the adverse health consequences of poor air quality may be related to generating oxidative stress (OS) in the lungs.
Air pollution contains particulate matter (PM) and oxidant gases, which could contribute to OS. A relatively recently discovered combustion product that could contribute to OS are environmentally persistent free radicals (EPFRs). 14 The term Environmentally Persistent Free Radical sounds like an oxymoron. Free radicals are generally short-lived, rapidly oxidizing whatever they contact within seconds. However, EPFRs persist in both the environment and in biological systems for prolonged periods; with a half-life of 21 days on PM2.5 15 and up to 5 years on wildfire charcoal. 16 EPFRs form readily in the post-flame and cool-zone regions of combustion systems and other thermal processes. EPFRs participate in the redox cycle in biological systems through the Fenton reaction and produce reactive oxygen species (ROS) over prolonged periods of time. 17 EPFR exposure increases susceptibility to influenza virus infection in mice, 18 induces OS in human epithelial cells in culture, 19 and is associated with wheeze outcomes in children. 20
The present study was undertaken to determine whether EPFR exposure increased the susceptibility of primary human nasal epithelial cells (NECs), grown into a fully differentiated respiratory epithelium at the air-liquid interface (ALI), 21 to infection with SARS-CoV-2. We hypothesized that EPFR exposure would increase susceptibility by reducing epithelial integrity and inducing OS. We further hypothesized that this effect could be prevented by pretreating the epithelium with an antioxidant.
Materials and methods (full methods are available in the online supplement)
Healthy non-atopic adults, aged 18 to 65 years, were recruited (Ethics approval: UQ2017000520; HREC61894; UQ2020001742).
Cell cultures
Primary NECs were collected, harvested, and cultured as previously described. 21 Briefly, nasal cells were scraped from the anterior surface of the inferior turbinate using a Rhino-Pro Nasal Curette (Arlington Scientific, UT, USA), grown as submerged cultures for approximately 2 weeks, “air-lifted” into ALI conditions, and maintained for at least 3 weeks until fully differentiated as verified by observing beating cilia by light microscopy and a high transepithelial electrical resistance (>1000 Ω/cm2). 22 Staining confirmed the presence of basal cells, goblet cells, ciliated cells, and tight junctions in the epithelium (Figure S1, OLS).
EPFR exposure
1,2-dichlorobenzene (DCB230) EPFRs (1 mg/cm2) were directly applied to the apical surface of the epithelium in ALI culture and left for four hours before removal.
Antioxidant pretreatment
Selected culture wells were pretreated with astaxanthin (20 µM) added to the basal media for 24 hours prior to other exposures. Astaxanthin was added with basal media changes every 24 hours to maintain an effective concentration throughout the experiments.
SARS-CoV-2 infection
NECs were infected with mock (PBS) or SARS-CoV-2 (1.25 × 105 plaque-forming units (PFU) in 100 µL) placed in the apical chamber, incubated for one hour at 37ºC. Basal media was changed every 24 hours and samples collected at 24, 48, and 72 hours post-infection. Cells were lysed with Buffer RLT containing 0.01% β-mercaptoethanol for RNA extraction or in protein lysis buffer (2% SDS/PBS, PhosSTOP, protease inhibitor) for protein extraction. All the procedures with active SARS-CoV-2 virus were performed under physical containment 3 (PC3) conditions and were approved by the University of Queensland Biosafety Committee (IBC/374B/SCMB/2020).
RNA isolation and reverse transcription‑quantitative polymerase chain reaction (RT‑qPCR) analysis
Total RNA was isolated from cells and reverse transcribed into cDNA. Quantification of gene expression was performed using a ViiA™ 7 Real-Time PCR System (Applied Biosystems, MA, USA).
Viral replication
Viral replication was quantified by measuring SARS-CoV-2 nuclear protein by western blotting.
Western Blot
Equal quantities of protein were loaded to Bolt™ 4–12%, Bis-Tris Gel (Thermo Fisher Scientific, MA, USA), electrophoresed at 200 V for 30 minutes, transferred to Immobilon-P PVDF membrane (Merck KGaA, Germany). Membranes were treated with relevant blocking buffers for one hour at room temperature and incubated with primary antibodies in blocking buffer at 4°C overnight. The following day, membranes were incubated with appropriate fluorescent secondary antibodies at room temperature for one hour, detected using the LI-COR Odyssey (BioAgilytix, NC, USA) and quantified by ImageJ (National Institutes of Health, Bethesda, MD).
PLpro activity
Inhibition of the SARS-CoV-2 papain-like protease (PLpro) enzymatic activity was measured according to the manufacturer’s instructions (E33972; Invitrogen™, CA, USA).
Statistical analysis
Data are presented as mean (SD) or median (25%–75%). Paired t-tests or sign-rank tests were used to compare data between experimental and control conditions. Kruskal-Wallis rank sum test with Dunn’s multiple comparison test was used to test comparisons between groups over time. Analyses were conducted using Stata/SE version 17.0 (StataCorp LLC, College Station, TX, USA) and statistical significance accepted at p < 0.05.
Results
SARS-CoV-2 infection caused an increase in viral replication in NECs, as indicated by SARS-CoV-2 nucleoprotein (NP) levels, at 24, 48, and 72 hour post-infection (overall p < 0.001) (Figure 1, Table S1). EPFR exposure prior to viral infection increased the NP levels (Table S1) (overall p < 0.001, compared with SARS-CoV-2 infection group (Figure 1(B)). Astaxanthin inhibited SARS-CoV-2 NP levels (p = 0.003, compared with SARS-CoV-2 alone) as well preventing the EPFR-induced increase of SARS-CoV-2 NP levels (p < 0.001, compared with EPFRs + SARS-CoV-2).
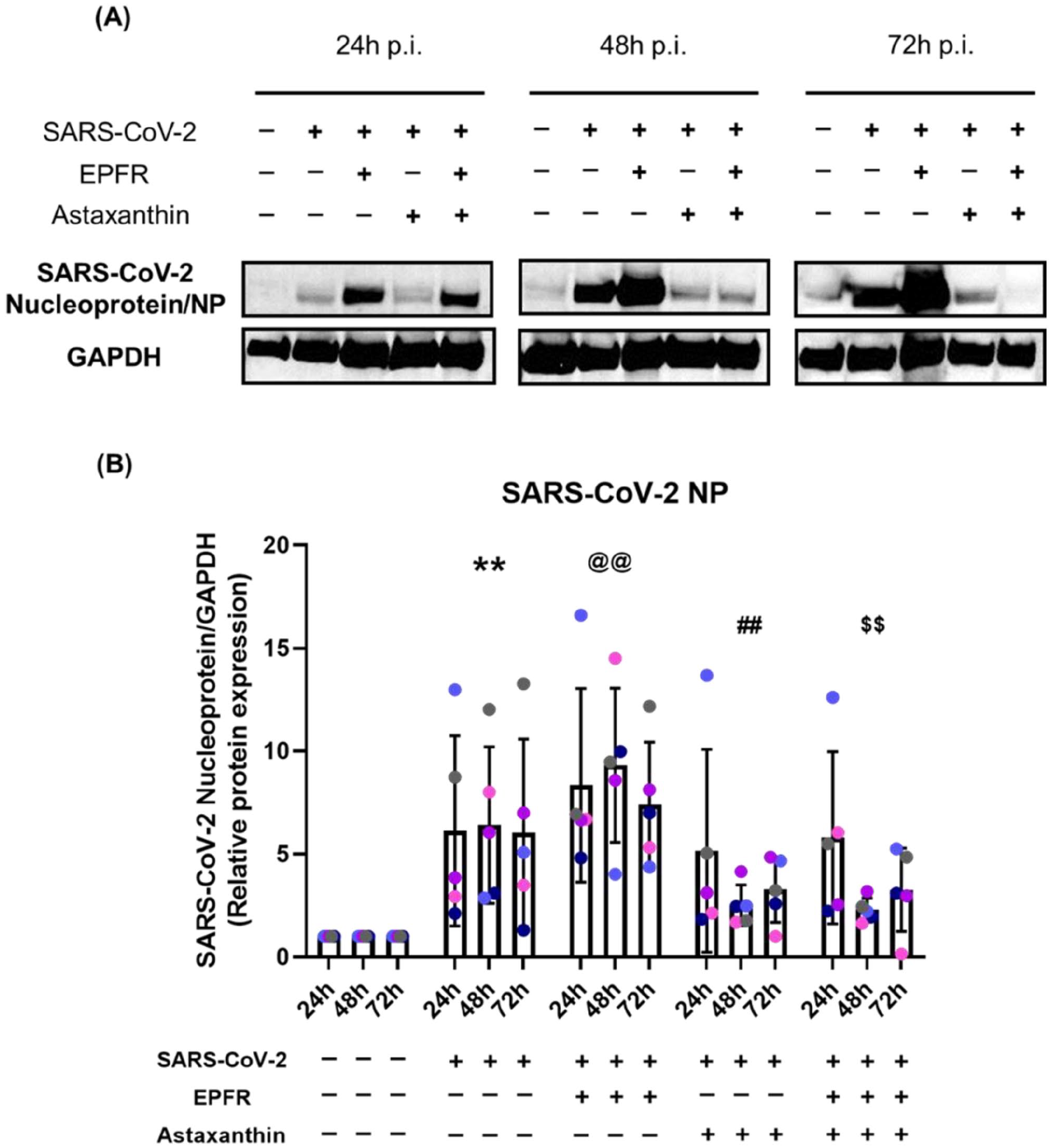
SARS-CoV-2 viral replication was increased by EPFR exposure but reduced by astaxanthin pretreatment. The well-differentiated airway epithelial cells were harvested at 24, 48, and 72 hours post-infection. Western blot images of SARS-CoV-2 nucleoprotein are shown from a representative individual (Panel A). SARS-CoV-2 nucleoprotein expression (NP), relative to GAPDH, for the group is shown in Panel B. NP expression was increased following infection with SARS-CoV-2 (**p < 0.001, compared with control) and further increase by EPFR exposure (@@ p = 0.003, compared with SARS-CoV-2 alone). Astaxanthin pretreatment decreased SARS-CoV-2 NP expression (##p = 0.003, compared with SARS-CoV-2 alone) and prevented the EPFR-induced increase in NP expression ($$p < 0.001, compared with EPFRs + SARS-CoV-2). Data presented as mean ± SD (n = 5 for each comparison). A single color was used in all figures to represent a single donor.
The spike protein of SARS-CoV-2 binds to angiotensin-converting enzyme (ACE) 2 receptor on NECs, facilitated by the transmembrane serine protease 2 (TMPRSS2). SARS-CoV-2 infection alone did not influence the expression of these surface receptors; however, exposure to EPFRs prior to infection upregulated expression of TMPRSS2 (p < 0.001), but not ACE2 (p = 0.85) (Figure 2, Tables S2&S3). Pretreatment with astaxanthin prior to infection downregulated expression of ACE2 (p < 0.001), had no effect on TMPRSS2 expression (p = 0.63), and prevented the EPFR-induced upregulation of TMPRSS2 (p < 0.001).
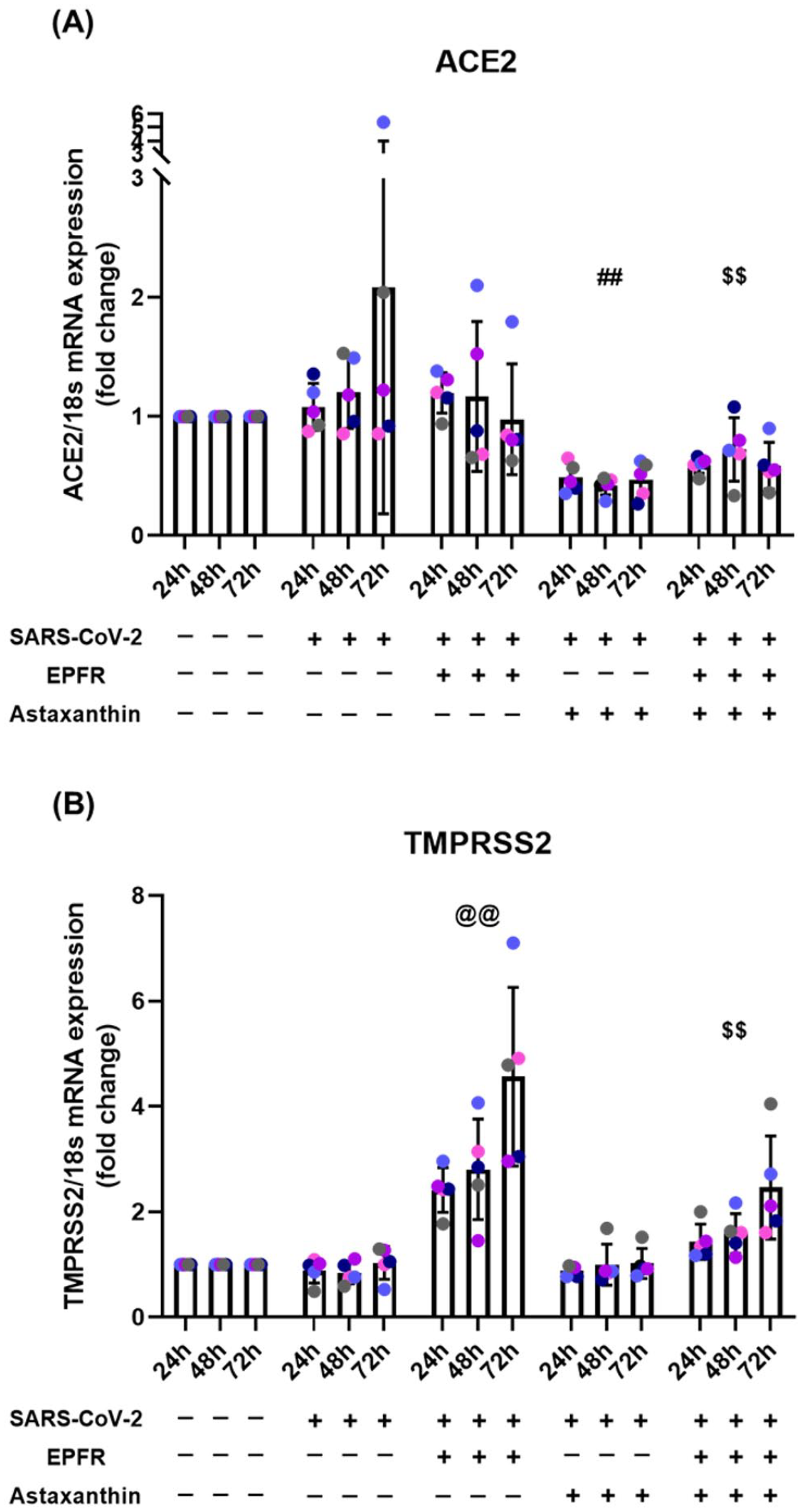
Impact of EPFR exposure and astaxanthin pretreatment on (A) ACE2 and (B) TMPRSS2 mRNA expression following epithelial infection with SARS-CoV-2. EPFR exposure increased expression of TMPRSS2 (@@p < 0.001, compared with SARS-CoV-2 alone) but not ACE2 (p = 0.85, compared with SARS-CoV-2 alone). Astaxanthin pretreatment prior to SARS-CoV-2 infection downregulated expression of ACE2 (## p < 0.001, compared with SARS-CoV-2 alone), but not of TMPRSS2 (p = 0.63, compared with SARS-CoV-2 alone). Astaxanthin pretreatment prior to EPFR exposure prevented the increase in TMPRSS2 expression ($$p < 0.001, compared with EPFRs + SARS-CoV-2). Data presented as mean ± SD (n = 5 for each comparison). A single color was used in all figures to represent a single donor.
SARS-CoV-2 infection caused an increase in cell death (p = 0.012, compared with control). EPFR exposure did not significantly increase cell death further (p = 0.23, compared with SARS-CoV-2 alone). Astaxanthin pretreatment had no effect on SARS-CoV-2 infection induced cell death (p = 0.42, compared with SARS-CoV-2 alone), but inhibited EPFRs and SARS-CoV-2 increased cell death (p = 0.040, compared with EPFRs + SARS-CoV-2) (Figure 3(A), Table S4).
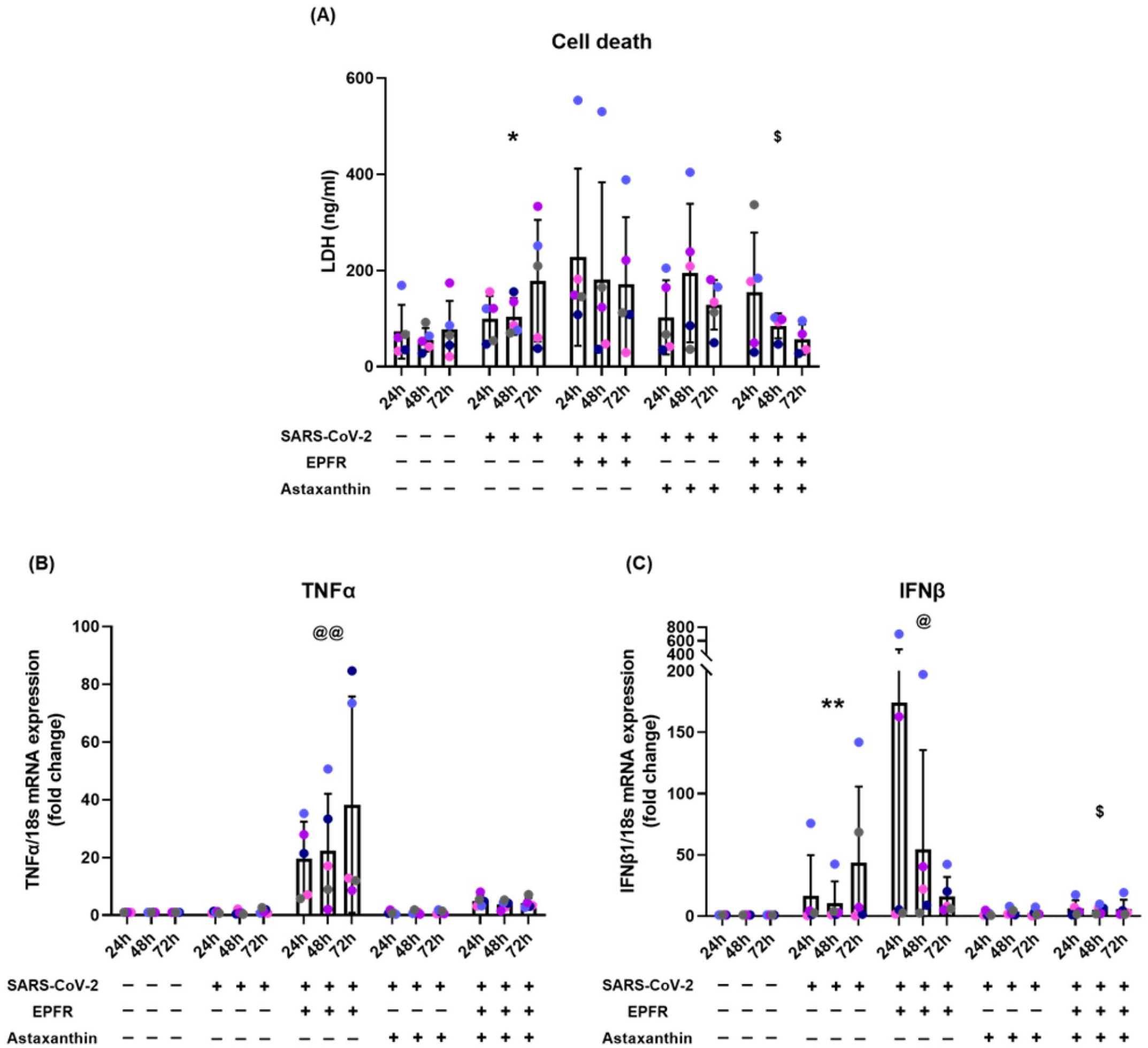
EPFR-induced increases in cell death, TNF-α, and IFN-β mRNA expression were prevented by astaxanthin pretreatment. Panel A shows cell death assays. SARS-CoV-2 infection caused an increase in cell death (*p = 0.012 compared with control), EPFR exposure did not further increase cell death (p = 0.23). EPFRs and SARS-CoV-2 induced cell death was prevented by astaxanthin ($p = 0.040, compared with EPFRs + SARS-CoV-2). TNFα mRNA expression is shown in Panel B. SARS-CoV-2 infection alone did not upregulate TNFα mRNA expression (p = 0.35). EPFR exposure significantly upregulated TNFα mRNA (@@p < 0.001, compared with control or SARS-CoV-2 alone). This increase was prevented by astaxanthin pretreatment (p = 0.063, compared with EPFRs + SARS-CoV-2). IFNβ mRNA expression is shown in Panel C. Expression was increased by SARS-CoV-2 infection (**p < 0.001, compared with control), further increased by EPFR exposure (@p = 0.017 compared with SARS-CoV-2 alone) and prevented by astaxanthin pretreatment ($p = 0.041, compared with EPFRs + SARS-CoV-2). Data presented as mean ± SD (n = 5 for each comparison). A single color was used in all figures to represent a single donor.
SARS-CoV-2 infection alone did not affect TNF-α mRNA expression (p = 0.35) (Figure 3(B)). EPFR exposure prior to SARS-CoV-2 infection progressively upregulated TNF-α mRNA expression (p < 0.001, compared with control or SARS-CoV-2 alone). Astaxanthin pretreatment prevented this increase, reducing TNF-α almost back to that induced by SARS-CoV-2 alone (p = 0.063, compared with EPFRs + SARS-CoV-2) (Figure 3(B)). Full data are show in the online supplement in Table S5.
SARS-CoV-2 infection alone progressively increased IFN-β mRNA expression (p < 0.001, compared with control) and EPFR exposure increased this further (p = 0.017, compared with SARS-CoV-2 alone). In addition, EPFR exposure resulted in early expression of IFN-β mRNA, peaking at 24 hours post-infection. Astaxanthin pretreatment had no significant effect on SARS-CoV-2 infection induced expression of IFN-β mRNA (p = 0.11, compared with SARS-CoV-2 alone) but inhibited the EPFR-induced increased IFN-β mRNA expression (p = 0.041, compared with EPFRs + SARS-CoV-2) (Figure 3(C)). Full data are show in the online supplement in Table S6.
To determine the impact of SARS-CoV-2 infection and EPFR exposure on cell stress pathways, mRNA expression of the following genes was examined: SIRT1-FOXO3 (antioxidant); p21 (apoptosis); PINK1 (mitophagy); and MUC5AC (mucin). SARS-CoV-2 infection alone did not have any effect on these genes (Tables S7 to S11). EPFR exposure prior to SARS-CoV-2 infection resulted in upregulation of p21 (p < 0.001, Table S9) and PINK1 (p < 0.001, Table S10) but marked downregulation of MUC5AC expression (p < 0.001) (Figure 4, Table S11). No effects were seen on SIRT1-FOXO3 expression (Tables S7 and S8). Pretreatment with astaxanthin prevented the EPFR-induced increase in p21 expression (p = 0.018, Table S9) but had no effect on EPFR-induced changes in PINK1 (p = 0.59, Table S10) and further reduced MUC5AC (p = 0.001, Figure 4, Table S11). Pretreatment with astaxanthin before SARS-CoV-2 infection upregulated SIRT1 and PINK1 expression (p < 0.001, Tables S7 and S10).
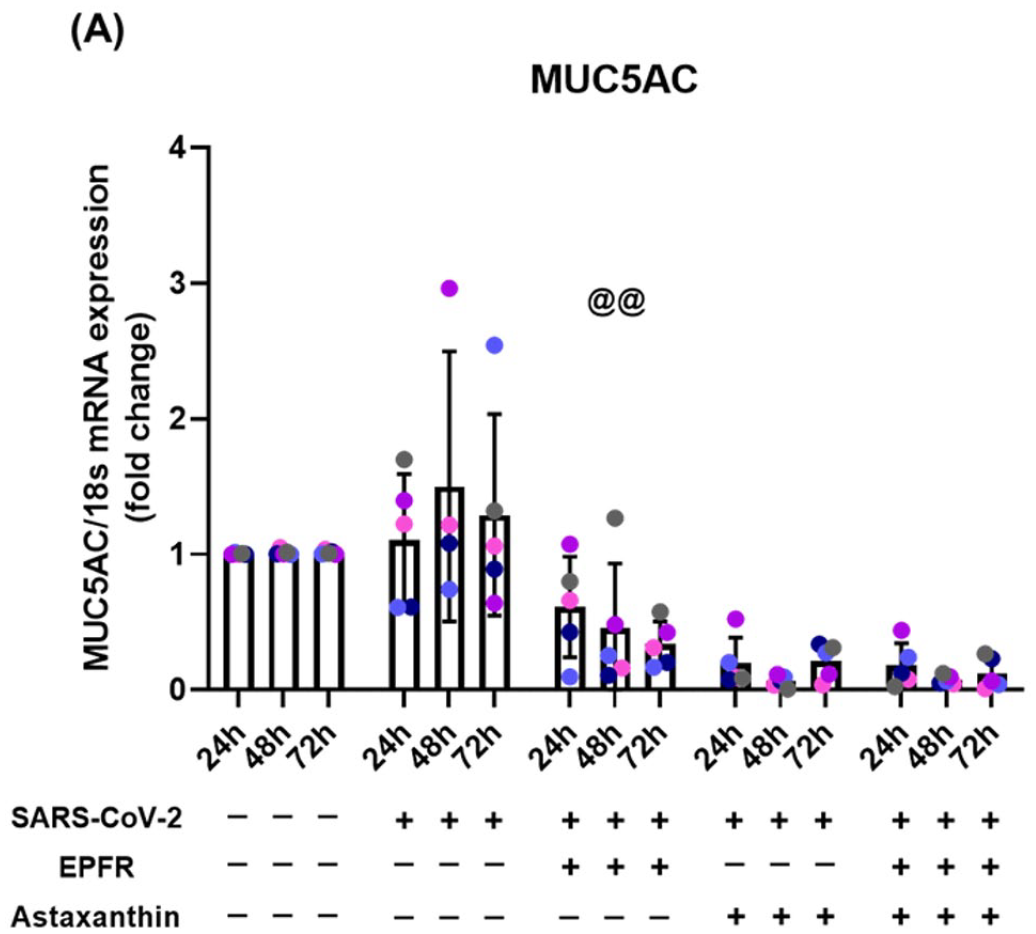
Reduction in MUC5AC mRNA expression following EPFR exposure that is not prevented by astaxanthin pretreatment. MUC5AC mRNA expression was not altered by SARS-CoV-2 infection alone (p = 0.33). EPFR exposure prior to SARS-CoV-2 infection resulted in decreased MUC5AC mRNA expression (@@p < 0.001, compared with SARS-CoV-2 alone). This decline was not prevented by astaxanthin pretreatment, but further downregulation was seen (p < 0.001). Data presented as mean ± SD (n = 5 for each comparison). A single color was used in all figures to represent a single donor.
The increase in SARS-CoV-2 replication and increased cytokine secretion induced by EPFR exposure and inhibited by astaxanthin pretreatment suggested that OS pathways may have been involved. To investigate this further, we examined the expression of the antioxidant enzymes catalase, SOD1, and TRX at the protein level. SARS-CoV-2 infection alone did not have a significant effect on catalase, SOD1, and TRX (p = 0.60, p = 0.13, p = 0.44, respectively, compared with control) (Figure 5). EPFR exposure prior to SARS-CoV-2 infection caused a decrease in SOD1 (p = 0.005 compared with control, p = 0.023 compared with SARS-CoV-2 alone) and TRX (p = 0.032 compared with control, p = 0.046 compared with SARS-CoV-2 alone) but not in catalase (p = 0.16 compared with control; p = 0.14 compared with SARS-CoV-2, Figure 5). Astaxanthin pretreatment returned protein levels toward control for catalase, SOD1, and TRX (p = 0.59, p = 0.18, p = 0.15, respectively compared with control) (Figure 5). Full data are shown in the online supplement (Table S12).
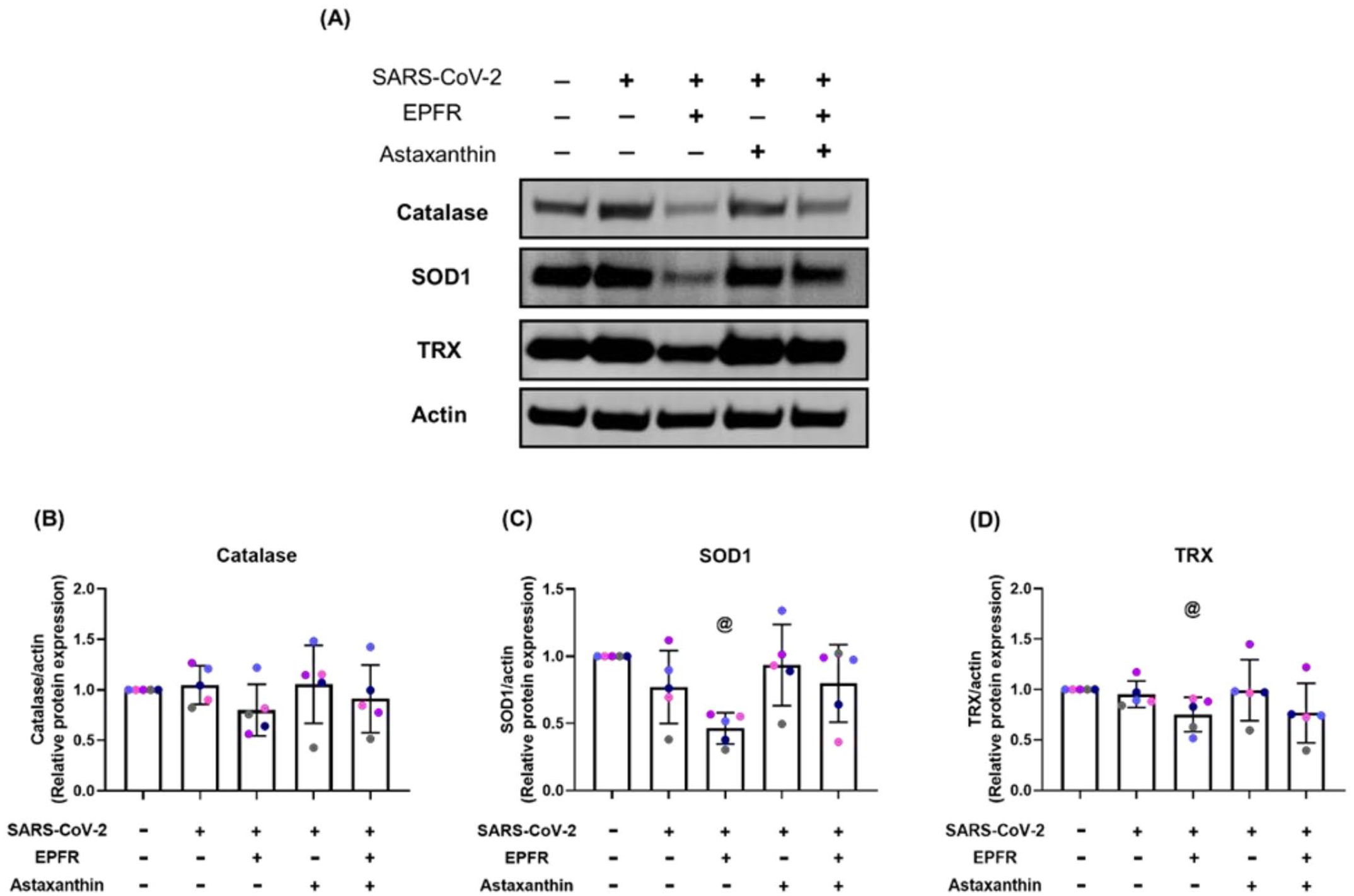
EPFR exposure prior to SARS-CoV-2 infection decreased oxidative stress defense enzyme proteins, with rescued by astaxanthin pretreatment. Panel A shows Western blot images of expression of catalase, superoxide dismutase 1 (SOD1), and thioredoxin (TRX) at the protein level from a representative individual. Group data are shown in Panels B, C, and D. SARS-CoV-2 infection alone did not alter enzyme expression. EPFR exposure prior to infection resulted in a non-significant decrease in catalase (p = 0.14) with significant decreases in SOD1 (@p = 0.023) and TRX expression (@p = 0.046) compared with SARS-CoV-2 alone. Astaxanthin pretreatment returned catalase, SOD1, and TRX levels toward control (p = 0.59, p = 0.18, p = 0.15, compared with control). Data presented as mean ± SD (n = 5 for each comparison). A single color was used in all figures to represent a single donor.
Discussion
Strong epidemiological evidence links poor air quality with increased susceptibility to SARS-CoV-2 infection, with increased case rates, severity, and mortality. Data from the present study show that prior exposure to EPFRs compromises epithelial integrity, increases SARS-CoV-2 replication, and induces a greater cytokine response from respiratory epithelial cells. EPFR exposure also markedly reduced expression of MUC5AC mRNA, the main gene involved in mucus production in the respiratory epithelium, and activated cell stress pathways, involved in apoptosis (p21) and mitophagy (PINK1). Most, but not all effects of EPFRs are likely to be secondary to the induction of OS in epithelial cells as they were prevented by pretreatment with the antioxidant astaxanthin. These data suggest this simple dietary antioxidant may improve outcome in those exposed to SARS-CoV-2 and that clinical trials are warranted.
We used EPFRs to investigate the effect of air pollution on SARS-CoV-2 infections. EPFRs are combustion products, formed in the post-flame and cool-zone regions of combustion systems, 14 and present on PM2.5 collected from polluted environments. 15 Chinese studies have estimated the personal EPFR exposure in polluted Beijing air to be equivalent to smoking more than five cigarettes per day 23 and demonstrated correlations between EPFR concentrations and ozone levels. 23 EPFRs participate in the redox cycle in biological systems through the Fenton reaction and produce ROS, with long-lasting effects in biological systems. 17 EPFRs induce OS in human respiratory epithelial cells in culture, increasing ROS generation, decreasing cellular antioxidant defenses, and inducing cell death. 19 In that model, addition of EPFR to PM2.5 increased cell death over that induce by PM2.5 alone. 19 Evidence that the EPFR-induced cell damage was related to OS came from data showing protection from cell damage was provided by pretreating cells with the antioxidant resveratrol. 19 EPFRs delivered to neonatal mice increased OS, increased susceptibility to respiratory viral infections, and increased airway responsiveness. 18 In that study, the damaging effects of EPFRs were prevented in mice overexpressing superoxide dismutase to enhance antioxidant capacity.
In the present study, we grew primary human NECs into a well-differentiated epithelium, complete with beating cilia and mucus producing goblet cells. SARS-CoV-2 infection of the epithelium resulted in a mild phenotype, with an increase in cell death and increase in IFN-β mRNA expression. However, no increase was seen in expression of the proinflammatory cytokine TNF-α mRNA, and no effects on cellular stress pathways were observed. EPFR exposure prior to SARS-CoV-2 infection damaged the epithelium in ways likely to increase susceptibility to viral infections. The reduced MUC5AC expression would decrease the protective mucus layer allowing easier viral access to cell surfaces. In addition, the increase in epithelial cell death seen following EPFR exposure would further increase viral access. In the present study, we showed an increase in viral replication in epithelial cells. These data are consistent with the increase in positive tests for SARS-CoV-2 following short-term exposure to increased levels of air pollutants which was reported in epidemiological studies.2,5 The upregulation of proinflammatory cytokine production and cell stress pathways, together with the reduction of antioxidant capacity following EPFR exposure we demonstrated, is consistent with the reports of increased severity and mortality from COVID-19 reported with chronic exposure to poor air quality.1,3,4
SARS-CoV-2 enters respiratory epithelial (and other) cells by binding the spike protein (S) to the ACE2 surface receptor, followed by acid-dependent proteolytic cleavage of S by enzymes including TMPRSS2. 24 Children are reported to express less ACE2 on nasal epithelial cells, 25 but whether this translates into a lower risk of severe COVID-19 is unclear. Higher expression of TMPRSS2 mRNA in nasal secretions of patients with COVID-19 is associated with increased respiratory distress. 26 Increased TMPRSS2 would be expected to increase SARS-CoV-2 entry into NECs, facilitating increased viral replication and increasing disease severity. In the present study, EPFR exposure prior to SARS-CoV-2 infection was associated with increased viral replication in NECs. Our data suggest EPFRs may increase viral replication by at least two mechanisms, decreasing epithelial integrity and increasing expression of TMPRSS2. Both these effects appear to be secondary to EPFR-induced OS as both are reduced by astaxanthin pretreatment.
Previous studies have used the antioxidant resveratrol to protect against the oxidant capacity of EPFRs. 19 In the present study, we chose to use astaxanthin as an antioxidant. Astaxanthin (3,3’-dihydroxy-β,β’-carotene-4,4’-dione) is a xanthophyll carotenoid found in various microorganisms and marine animals. 27 Astaxanthin is a lipophilic compound that spans the cell membrane and provides protection against free radicals and other oxidant stressors.28,29 As reviewed by Kidd, 29 in spanning the cell membrane astaxanthin provides antioxidant protection by multiple mechanisms, including neutralizing free radicals by donating electrons to unpaired electrons or by capturing unpaired electrons; bonding with radicals to form non-reactive “adducts”; and removing electrons or electronic energy from the membrane. As astaxanthin carries very low net molecular energy it provides further protection via resistance to transformation into a pro-oxidant molecule. 29 Astaxanthin also protects cells from oxidant and inflammatory damage by inhibiting various intracellular pathways (reviewed in Kidd). 29 In the present study, astaxanthin pretreatment was able to prevent EPFR-induced degradation of epithelial integrity, increase in TNF-α mRNA expression, increase in p21 mRNA expression, and decrease in caspase expression. Whether this protection occurred by neutralizing the redox ability of EPFRs or via direct protective effects on the cells is unclear. However, astaxanthin was not able to prevent EPFR-induced reduction of MUC5AC mRNA expression, suggesting that simple radical neutralization may not be the entire story. Further research to clarify the potential of astaxanthin to protect people against the adverse effects of an oxidant environment is certainly warranted.
Perhaps the most surprising result from the present study was the ability of pretreatment with astaxanthin to reduce SARS-CoV-2 replication in epithelial cells. Two key enzymes involved in SARS-CoV-2 replication and transcription are main protease (Mpro) 30 and papain-like protease (PLpro).30,31 A previous in silico study demonstrated that astaxanthin has a high binding energy (−9.3 kcal/mol) to dock into the active site of PLpro and inhibit its activity. 32 Astaxanthin has less ability to bind and inhibit Mpro. 32 Whether this theoretical action of astaxanthin is the mechanism by which SARS-CoV-2 replication was inhibited in epithelial cells and whether this translates into clinical activity remains to be investigated. We demonstrated that astaxanthin can inhibit PLpro activity in vitro, but that at the dose we used (20 µM), PLpro activity was decreased by approximately 20% (Figure S2). Whether this reduction would be sufficient to inhibit SARS-CoV-2 replication is unknown and warrants further study.
The present study has some strengths and limitations. Strengths include growing primary human nasal epithelial cells into a fully differentiated respiratory epithelium to provide a physiologically accurate model. The nasal epithelium is an appropriate model to use as it represents the site of first contact and infection with respiratory viral infections, including SARS-CoV-2. Limitations include the small sample size we were able to study; the requirement to use PC3 conditions meant that materials had to be neutralized prior to removal from the facility which made some analyses, for example, enzyme function assays, impossible; and the fact that we could not examine contributions from inflammatory or immune cells in our study. In addition, we used the ancestral SARS-CoV-2 strain, which may limit generalizability to newer variants. Despite these limitations, the results from the present study strongly suggest that oxidant stress from pollution-associated EPFRs increased susceptibility to SARS-CoV-2 infection and that pretreatment with the antioxidant astaxanthin may provide protection. A comparison of SARS-CoV-2 infection and the impacts of EPFRs on SARS-CoV-2 infection is shown in Figure 6.
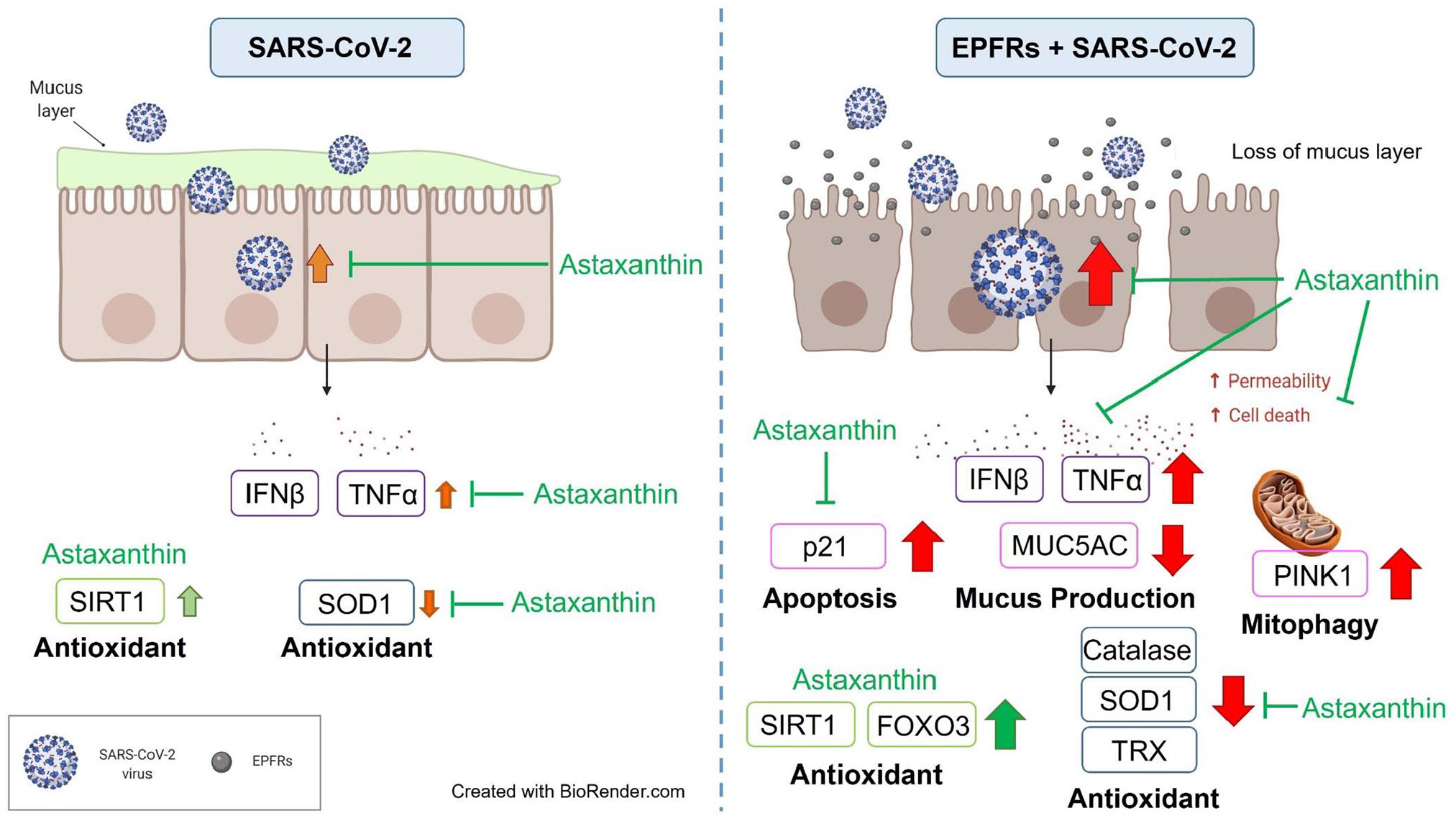
Comparison of SARS-CoV-2 infection and the impacts of EPFRs on SARS-CoV-2 infection. EPFR exposure increased susceptibility to SARS-CoV-2 infection with higher viral replication, higher cytokine secretion, and lower antioxidant enzymes compared with SARS-CoV-2 infection alone. The antioxidant, astaxanthin, protected the well-differentiated epithelium by inducing the antioxidant pathway, inhibiting the apoptosis, and increasing the antioxidant enzyme activities (red: negative impact; green: positive impact).
Supplemental Material
sj-pdf-1-ebm-10.1177_15353702221142616 – Supplemental material for Environmentally persistent free radicals enhance SARS-CoV-2 replication in respiratory epithelium
Supplemental material, sj-pdf-1-ebm-10.1177_15353702221142616 for Environmentally persistent free radicals enhance SARS-CoV-2 replication in respiratory epithelium by Ayaho Yamamoto, Peter D Sly, Keng Yih Chew, Lavrent Khachatryan, Nelufa Begum, Abrey J Yeo, Luan D Vu, Kirsty R Short, Stephania A Cormier and Emmanuelle Fantino in Experimental Biology and Medicine
Footnotes
Authors’ Contributions
Conception and design: AY, PDS, LDV, KRS, SAC, and EF; participant recruitment: AY and PDS; experimental performance: AY and KYC; EPFR production: LK; data analysis and interpretation: AY, PDS, AJY, and EF; statistical analysis: PDS and NB; manuscript preparation: AY, PDS, KRS, SAC, and EF. All authors reviewed and approved the final manuscript as submitted.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
The University of Queensland Human Research Ethics Committees: UQ2017000520; UQ2020001742. Children’s Health Queensland Hospital and Health Service Human Research Ethics Committee: HREC61894.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by a grant from the National Institute of Environmental Health Sciences (3P42 ES013648-08A1S1), and PDS is funded by the National Health and Medical Research Council, Australia (1193840).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.