
Abstract
Succinate dehydrogenase complex subunit C (SDHC) is a subunit of mitochondrial complex II (MCII), which is also known as succinate dehydrogenase (SDH) or succinate: ubiquinone oxidoreductase. Mitochondrial complex II is the smallest respiratory complex in the respiratory chain and contains four subunits. SDHC is a membrane-anchored subunit of SDH, which connects the tricarboxylic acid cycle and the electron transport chain. SDH regulates several physiological processes within cells, plays an important role in generating energy to maintain normal cell growth, and is involved in apoptosis. Currently, SDHC is generally recognized as a tumor-suppressor gene. SDHC mutations can cause oxidative damage in the body. It is closely related to the occurrence and development of cancer, neurodegenerative diseases, and aging-related diseases. Here, we review studies on the structure, biological function, related diseases of SDHC, and the mev-1 Animal Model of SDHC Mutation and its potential use as a therapeutic target of certain human diseases.
Keywords
Impact Statement
Much attention has been focused on oxidative damage that plays a role in cancer and cellular aging. SDHC as a subunit of mitochondrial complex II plays an important role in generating energy to maintain normal cell and organismal growth. Here, we provide an insight into the currently available knowledge of the structure and biological function of SDHC, including its relationship to various diseases, and discuss whether SDHC might represent a future therapeutic target.
Introduction
The succinate dehydrogenase complex subunit C (SDHC) protein is a large subunit with cytochrome b in mitochondrial complex II, a small peptide with 15.5 kDa. The SDHC structure has 140 amino acids and contains five α Helix and three transmembrane segments. The active gene SDHC has six exons and five introns, extending over 35 kb and is located at position 1q21, adjacent to the pericentric heterochromatin on the long arm of chromosome 1. 1 SDHC is the membrane anchor of mitochondrial complex II (MCII, or succinate dehydrogenase [SDH] or succinate: ubiquinone oxidoreductase [SQR]). 2 A genome sequence analysis indicated that SDHC was a housekeeping gene. SDHC exists in at least two protein complexes in the inner mitochondrial membrane and plays important roles in electron transfer and protein assembly. 3 SDHC dysfunction caused by oxidative stress contributes to the occurrence of some diseases. The specific mechanism of SDHC involvement in the occurrence or development of tumors or other diseases is unclear. An in-depth understanding of the biological function of SDHC and its mechanism of action in related diseases may help identify clinical therapeutic targets. Here, we review studies on the biological function of SDHC and diseases caused by SDHC dysfunction and discuss the possible research directions for using SDHC as a therapeutic target.
Physiological functions of SDHC
Participation in MCII assembly
ATP synthesis in aerobic eukaryotes mainly occurs in mitochondria through oxidative phosphorylation. Electrons are added to oxygen molecules by four complexes, including MCII (SDH or SQR) connecting the tricarboxylic acid cycle and the oxidative phosphorylation process. MCII contains four subunits: two hydrophilic and two hydrophobic. The flavoprotein (SDHA) and iron–sulfur protein (SDHB) subunits form the hydrophilic catalytic center of SDH inserted into the mitochondrial matrix. SDHC and SDHD are the hydrophobic transmembrane subunits that form the membrane-anchoring domain embedded in the inner mitochondrial membrane. There is a heme and a ubiquitin-binding site between SDHC and SDHD (Figure 1(a)). 4 SDHA and SDHB contain two types of prosthetic groups: flavin adenine dinucleotide (FAD) and iron–sulfur clusters. Stabilization of the SDHC–SDHD dimer requires a hydrophilic domain, and deletion of SDHA or SDHB results in an almost complete loss of the dimer. 5 The SDHA–SDHB dimer can exist without one or both membrane anchors. 6 This indicates that the SDHA–SDHB dimerization forming an active hydrophilic structure may precede the SDHC–SDHD dimerization during SDH assembly. In addition, heme b contributes to the structural integrity of the membrane-anchoring domain. 7 However, the process of SDHC–SDHD dimerization during SDH assembly is poorly understood.
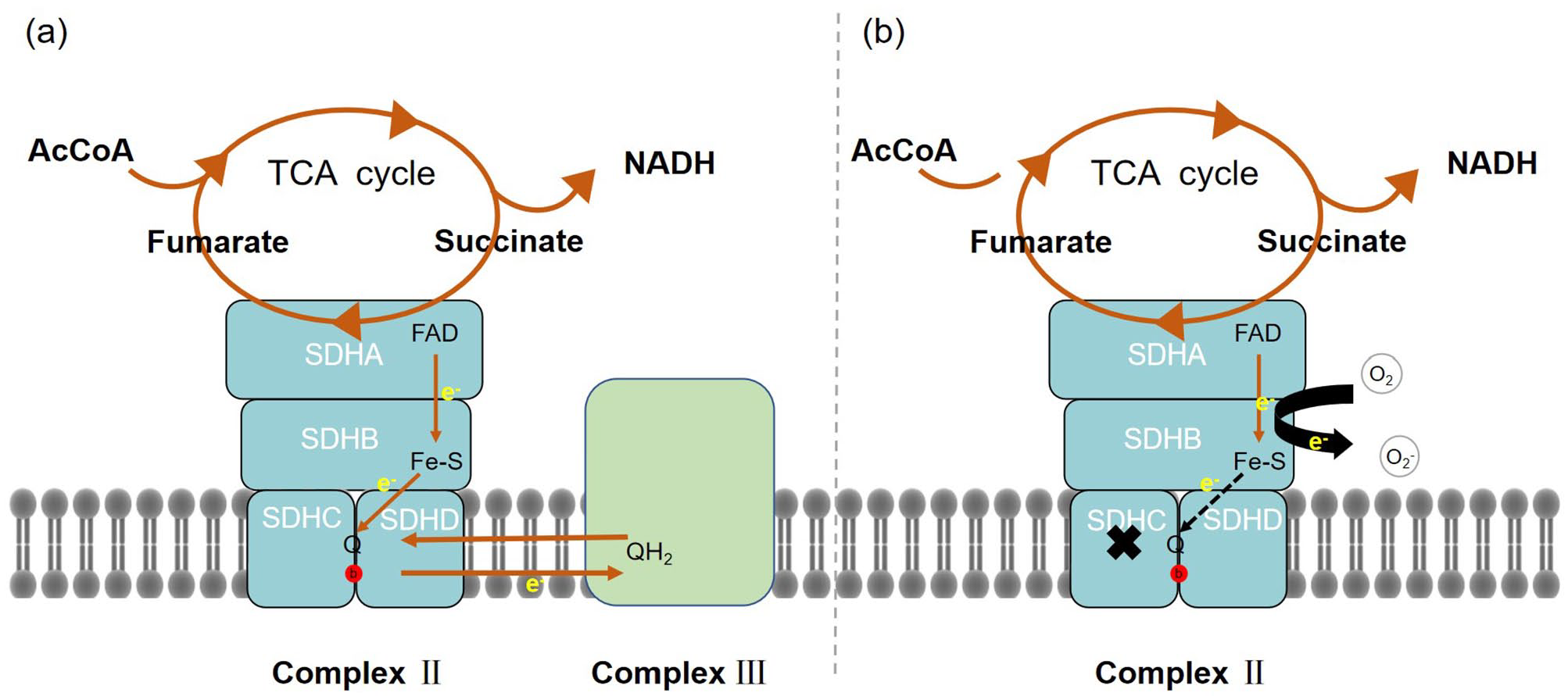
SDH participates in the mitochondrial electron transport chain and tricarboxylic acid cycle in the mitochondrial matrix. (a) Hypothetical model of SDH compound under normal function, which contains four subunits, that is SDHA, SDHB, SDHC, and SDHD. The red circle represents heme. (b) A hypothetical model of SDHC subunit inactivation, which contains four subunits, that is, SDHA, SDHB, SDHC, and SDHD. The red circle represents heme.
In the presence of SDHC mutations or the absence of normal SDHC, SDHA and SDHB are imported into mitochondria but do not successfully assemble into MCII. 8 SDHC downregulation reduces the activities of MCII and mitochondrial complex IV (MCIV) but does not affect respiratory body levels or cell numbers. 9 This finding rules out the involvement of MCII, particularly SDHC, in the respiratory body. However, MCII may play a role in maintaining respiratory body integrity. In addition, because all other complexes in the respiratory chain contain some polypeptide subunits encoded by the mitochondrial genome, and all four subunits of MCII are completely encoded by nuclear genes, respiratory chain defects in MCII are relatively rare.
Involved in electron transfer in respiratory chain
Owing to different roles in the reaction process, MCII has two distinct enzymatic activities: SDH and SQR. SDH activity is completed by the dimer of SDHA and SDHB. In the tricarboxylic acid cycle (TCA), coenzyme Q (CoQ) acts as an electron acceptor, oxidizing succinic acid to fumaric acid. The electrons generated in this process are transferred from the succinic acid-binding site of the substrate SDHA subunit to the iron sulfur center of the SDHB subunit, and finally to the ubiquinone-binding site (QP). Contrary to the two-electron reduction of FAD, ubiquinone reduction occurs in two stepwise one-electron reactions. The QP site significantly stabilizes the partially reduced semiquinone for the complete reduction of ubiquinol.10,11 QP is formed by SDHB, SDHC, and SDHD subunits of the intima. 12 In addition, it was found in Escherichia coli that heme b, located between SDHC and SDHD, plays a key role in the energy transfer process. 13
The mev-1 mutation of Caenorhabditis elegans encoding SDHC subunit homolog did not affect the SDH activity in mitochondria, but it reduced the MCII activity of mitochondrial membrane by more than 80%, which led to the separation of the two enzyme activities of MCII, the SQR activity of MCII was reduced, and the SDH activity was not affected. It shows that SDHC is directly involved in the electron transfer from MCII to CoQ. 14 The absence of SDHC affects the formation of the enzyme complex, destroys its stability, and damages its function, resulting in the occurrence of disease. SDHC mutation may affect the coenzyme Q-binding and heme b sites at the junction of SDHC and SDHD (Figure 1(b)). QP is an important source of reactive oxygen species (ROS). 15 In complex III, the function of cytochrome b is to remove hemiquinone by effectively acting as Q dismutase in the original exercise Q cycle. 16 Cytochrome b560 in complex II may also have a similar function in stabilizing or decomposing semiquinone, and SDHC mutation may eliminate this function, thus making semiquinone accumulate. Its reaction with oxygen will lead to excessive production of superoxide. SDHC mutation may increase the production of superoxide in two ways. They may allow more electrons to leak from the MCII. The electrons leaked from the respiratory chain undergo one-electron reduction with molecular oxygen to generate a superoxide anion, which is the main source of ROS in the body. Subsequently, oxidative stress occurs. ROS contributes to normal physiological processes of cells at low concentrations but causes cell damage at high concentrations. 17
Formation of the TIM22 complex
The SDH3 assembly with two distinct partner proteins, that is, SDH4 and Tim18, is recruited to two distinct mitochondrial membrane complexes with roles in bioenergetics and protein biosynthesis, respectively. In addition to being MCII subunits, Sdh3 and Tim18 participate in the biosynthesis and assembly of Tim22 and Tim54 into a functional TIM22 complex (Figure 2). 3 The TIM22 complex acts as a carrier translocase to facilitate the insertion of a class of hydrophobins with internal targeting signals into the inner mitochondrial membrane and plays an important role in protein assembly. 18
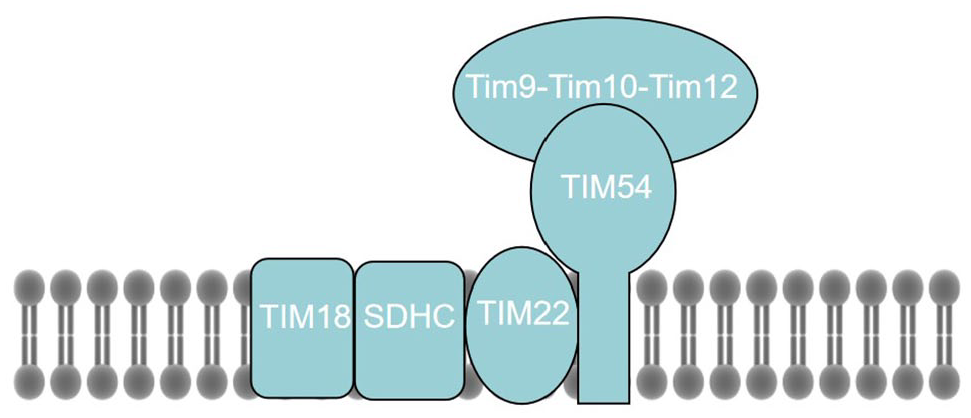
Hypothetical model of the TIM22 complex, which contains three membrane-integral subunits, that is, Tim54, Tim22, and Tim18, and the peripheral chaperone complex Tim9–Tim10–Tim12.
Promotion of apoptosis
SDH is an apoptosis sensor, 19 and SDHC is a tumor-suppressor and pro-apoptotic gene. 20 SDHC overexpression exerts a strong pro-apoptotic effect that is pH-dependent. As pH decreases, SDHA and SDHB dissociate from membrane-bound components of MCII required for SQR and SDH activities. Apoptosis is not induced by simultaneous inhibition of SQR and SDH activities but by reduced SQR activity alone, SDH activity is not affected, while superoxide is produced and cell apoptosis is promoted. 21 The efficiency and kinetics of pro-apoptotic effects of SDHC overexpression suggest that it may directly affect the MCII assembly. 3 SDHC overexpression induces apoptosis accompanied by a transient decrease in MCII activity and production of oxygen radicals. 22 Cells constitutively deficient in SDHC are resistant to the effects of various pro-apoptotic cytostatic drugs and Fas receptors. Inactivation of MCII subunits in cancer cells may compromise MCII as a whole, thus inhibiting SDH and SQR activities of MCII simultaneously, rendering MCII unable to induce apoptosis or promote tumorigenesis. 21
Related diseases
SDHC mutation leads to excessive ROS production in cells. Excessive electron leakage from the mitochondrial respiratory chain converts oxygen molecules to a superoxide anion in mitochondria that quickly converts to hydrogen peroxide, which undergoes Fenton’s reaction to convert into a highly reactive hydroxyl radical. Superoxide anion, hydrogen peroxide, and hydroxyl radicals are collectively referred to as ROS. Destruction of cellular components by oxidative stress leads to many lifestyle and age-related diseases, such as diabetes and arteriosclerosis.23 –25 Loss of the subunit function of SDH usually does not lead to a single common pathology but to various phenotypes of neurological diseases and tumors.
Neurodegenerative disease
The brain weighs only 2% of the body weight but consumes 20% of the total energy consumed by the body. The subunits and cofactors in MCII assembly play important roles in the nervous system, particularly in neurodegenerative diseases.26 –29 SDHC V69E-mutant mice have impaired mitochondrial electron transport chain function, chronic oxidative stress during physiological aging, and astrocyte defects accompanied by stress-activated protein kinases (SAPKs)/c-Jun N-terminal protein kinases (JNKs) activation and Ca2+ overload. 30 Astrocytes are cells that play an important role in neuronal metabolism. In addition, SDH activity supports the development and function of the nervous system. 31
SDHC mutation and inhibition of SDHC activity are closely related to the occurrence of neurodegenerative diseases. Neurons, particularly midbrain substantia nigra pars compacta (SNpc) dopaminergic neurons, are vulnerable to mitochondrial dysfunction because their energy comes from the mitochondrial glycolytic pathway or mitochondrial transfer between axons and dendrites. SDHD and SDHC bind to form the membrane-anchoring domain of SDH. In tyrosine hydroxylase-SDHD mice, in addition to reduced cell numbers in the adrenal medulla, carotid body, and superior cervical ganglia, dopaminergic neurons in postnatal SNpc were reduced. Furthermore, metamaturity was inhibited, and dopamine neuron cells were progressively lost within the first year of life. 32 SNpc is the most important neuronal population affected by Parkinson’s disease (PD), as mitochondrial damage is involved in the pathogenesis of this neurodegenerative disorder. Mitochondrial dysfunction is closely related to the occurrence and development of PD. Significant downregulation of SDHC was found in meta-analysis of substantia nigra and peripheral blood samples from patients with PD. 33 Significant downregulation of SDHC was also found in the study of differential genes between parkinsonians without PARK2 or PARK8 mutations and healthy individuals. 34 In addition, an omics study on SNpc tissue samples from α-synuclein transgenic mice revealed significant SDHC downregulation. 35
Although it is not clear how SDHC participates in the pathogenesis of PD, oxidative stress causes cell damage, DNA repair damage, and mitochondrial dysfunction, which has been considered as a common cause of neurodegenerative diseases. It is also known that SDH is a key enzyme complex in mitochondrial TCA cycle and aerobic respiratory chain. MCII deficiency and reduced activity are found in patients with neurodegenerative diseases, such as Alzheimer’s disease (AD) and PD.28,36,37 SDH-mediated pyruvate metabolism is a key metabolic reaction in cells that leads to mitochondrial ATP production and drives several other biosynthetic processes. Pyruvate metabolism is abnormal in both AD and PD, with elevated pyruvate levels in cerebrospinal fluid and serum, respectively. SDH binds indirectly to various signaling pathways, such as the mammalian target of rapamycin/regulatory element-binding protein pathway, which plays a crucial role in lipid synthesis. 38 Lipid metabolism disorder, mitochondrial damage, apoptosis, free radicals, and oxidative stress are involved in the mechanism of PD. Therefore, the role of SDHC as the membrane-anchoring domain of SDH in PD should be further studied.
Tumor
SDHC is a tumor-suppressor gene. It is a key player in the differentiation of cancer cells. SDHC has been implicated in the generation of pro-apoptotic signals. 22 In SDHC E69 cells, cytochrome c released from mitochondria was significantly increased; oxidative stress activates the p53 and Ras signal transduction pathways. 39 In addition, Hamster fibers expressing a mutation in SDH subunit C (SDHC; B9) increases the production of ROS, which can lead to metabolic stress, genomic instability, nuclear DNA damage, mutation, and tumorigenesis.40,41 The increased ROS production may activate hypoxia-inducible factors by simulating hypoxia signal pathways, thus promoting cell proliferation, angiogenesis, and clinically observed tumor phenotype. The tumorigenic potential of SDHC mutations has also been suggested to derive from the accumulation of succinate, which inhibits α-ketoglutarate-dependent prolyl hydroxylases (PHDs), thus causing a pseudohypoxia condition with hypoxia-inducible factor 1α (HIF-1α) stabilization and nuclear translocation in normoxia.42,43 This would result in constitutive activation of genes that favor tumor growth. 44 These findings suggest that ROS generated by mitochondrial damage in SDHC trigger tumor progression.
SDHC transcripts have a deleted alternative splicing site, and two alternative splicing variants (ASVs) are generated through an alternative splicing mechanism. ASVs have a dominant-negative effect on SDHC activity and are associated with human disease. ∆3 ASV lacks exon 3 and encodes the major region of MCII oxidoreductase activity, mainly affecting a part of the succinate coenzyme Q oxidoreductase. ∆5 ASV lacks the heme-binding domain because of a frameshift mutation and is defective in exon 5 encoding the heme b560-binding domain, which results in marked reduction in SDH complex activity. 45 This variant of SDHC may act as a 6/25 dominant-negative inhibitor of full-length SDHC. Since electrons cannot be transferred correctly, they leak easily to produce excess superoxide. These subtypes may play a role in SDH dysfunction-related tumorigenesis, including HCT-15 colorectal adenocarcinoma cells, where overexpressions of the Δ3 SDHC isoform and Δ5 ASVs are associated with decreased SDH activity and increased ROS production. 45 Aberrant GATA1S expression in myeloid leukemia alters differentiation and proliferation potentials of hematopoietic precursors and leads to a poor prognosis. Aberrant SDHC ASV expression enhances the leukemic potential of GATA-1S. 46 Alternatively spliced isoforms of SDHC may be involved in tumor cell differentiation. ASV has a dominant-negative effect on SDHC activity and causes incomplete inhibition of SDHC transcription. During tumorigenesis, alternative splicing mechanisms may already exist in cancer cells evolving from differentiated cell types. 47 Therefore, alternative splicing in SDHC tumor cells may reflect the pre-existing alternative splicing machinery in parental cells. An understanding of factors regulating alternative splicing of SDHC could aid in the pathogenesis of SDHC ASV production in tumors associated with SDH dysfunction.
Paraganglioma is a tumor that usually arises in neuroendocrine tissue along the paravertebral axis. Of all cases, 35% are hereditary, caused by mutations in subunits of MCII. Paraganglioma-3, first described by Niemann and Muller in 2000, is a paraganglioma syndrome of autosomal dominant inheritance, caused by SDHC mutations located at 1q21–q23.48,49 A case series of patients with SDHC mutations from the United Kingdom revealed SDHC mutations in head and neck paraganglioma, extra-adrenal paraganglioma, and pheochromocytoma. 50 Head and neck paragangliomas associated with SDHC mutations are almost always benign and mostly solitary. 51 A case of catecholamine-producing malignant paraganglioma due to SDHC mutation showed G→T transversion in intron 5 + 1 of SDHC, resulting in exon 5 deletion, reading frameshift, 52 and aberrant splicing. Exon 5 encodes part of the first transmembrane segment and the entire second transmembrane segment of SDHC. This may interfere with anchoring of the catalytic subunit of MCII to the mitochondrial membrane and, ultimately, enzyme failure.
Gastrointestinal stromal tumor (GIST) related to SDHC mutation, mainly originating from the stomach, the most frequent and greatest contiguous change involved loss of the chromosomal region 1q12–q23.3. Most patients with Carney triad (dual manifestation of GIST and paraganglioma) have tumors that exhibit SDHC promoter hypermethylation. 53 DNA methylation changes are hallmarks of human cancer. 54 SDHC promoter hypermethylation is most common in SDH-deficient GISTs, present in up to one-third of cases, with Carney triad in 50% of cases.55,56
SDHX mutation is an intrinsic factor and the driving mechanism of epithelial–mesenchymal transition (EMT). 57 SDHC is a contributing factor in breast cancer. Its downregulation in breast cancer promotes EMT with concomitant structural remodeling of mitochondrial organelles. In a comprehensive analysis of a cohort of patients with breast cancer, inverse association between EMT and SDHC underexpression was relatively consistent and particularly pronounced in basal-like molecular cancer subtypes, worsening the patient’s prognosis. 58
Renal cell carcinoma originates in the kidney, and SDH-deficient renal carcinoma was introduced in the World Health Organization 2016 classification. The main pathological features are vacuolated eosinophilic cytoplasm and cytoplasmic inclusions. It is mainly associated with SDHB mutations, with fewer mutations in SDHC and SDHA. 59 Loss of SDH complex activity associated with SDHC in liver cancer plays a key role in promoting growth and metastasis of hepatocellular carcinoma mainly through the ROS/nuclear factor kappa B signaling pathway. 60
Central obesity
During the development of a familial pheochromocytoma and paraganglioma mouse model, Kang et al. incidentally found that SDHC-deficient tyrosine hydroxylase-expressing mice developed non-diabetic obesity in adulthood. SDHC deletion results in loss of dopaminergic function and catecholamine production and reduced dopaminergic signaling, which in turn affects the dopamine signaling pathway to regulate food intake and energy metabolism. 61 SDHC single-nucleotide polymorphisms are associated with the body mass index and the obesity risk. 62 Altered gene expressions in obese SDHC-deficient mice could explain the balance between the central nervous system and peripheral adrenergic deficits.
Emphysema
SDHC is also a key regulator of emphysema-driven skeletal muscle respiration and fatigue. It supports respiration of cultured muscle cells. Mice with emphysema showed downregulation of SDHC, but not of SDHA, SDHB, or SDHD in skeletal muscles and significantly reduced SDH activity. 63 SDHC can regulate the muscle fiber type independent of its SDH complex enzyme function. SDHC overexpression results in more type 2A and 2X fibers. It abrogates decreased oxygen consumption and fatigue tolerance in animal models of emphysema-induced skeletal muscle dysfunction. 64 This mechanism may explain SDH function in patients with chronic obstructive pulmonary disease and skeletal muscle dysfunction.
SDHC mutation model
Mev-1 (kn-1) mutant gene is homologous to SDHC in human MCII. At present, the mev-1 models studied include SDHC E69 cells, G71E in Caenorhabditis elegans, I71E in Drosophila melanogaster, and V69E in mice (Table 1).41,65
Functional changes of SDHC gene mutation biological model.
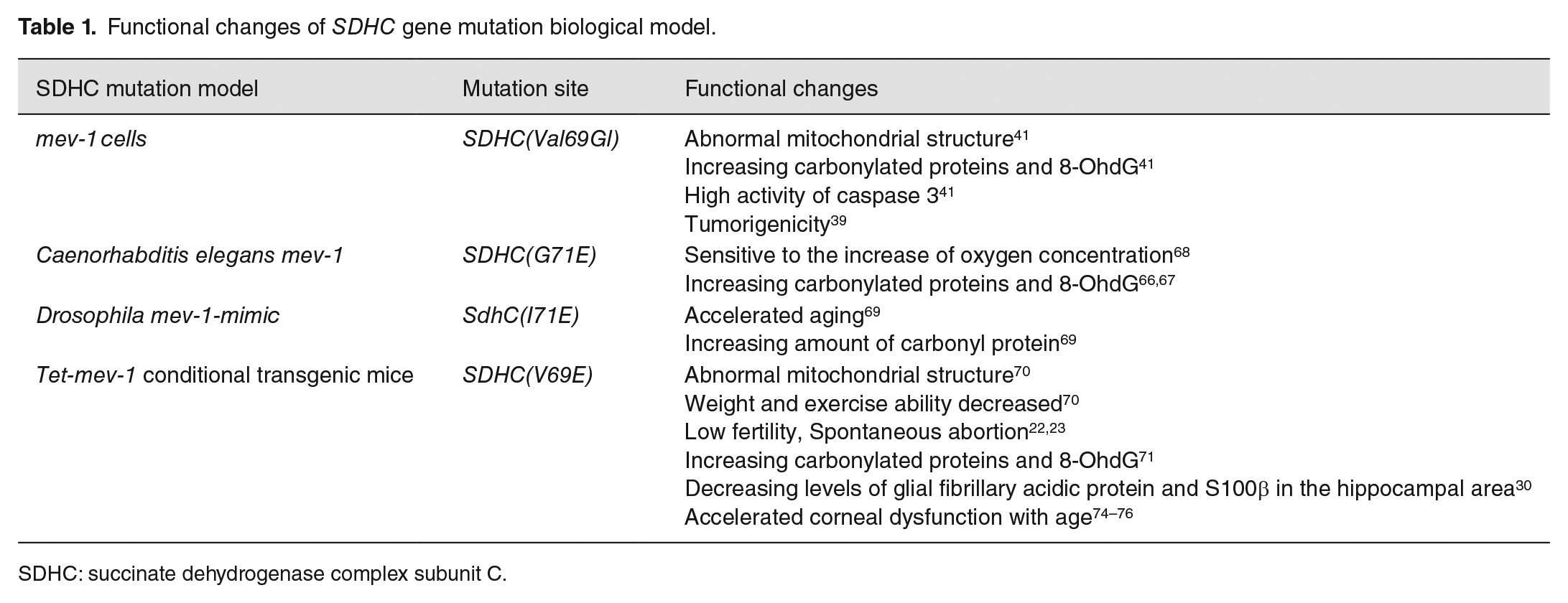
SDHC: succinate dehydrogenase complex subunit C.
In a transgenic mouse embryonic fibroblast NIH3T3 cell line, the mutation at the 69th position, changing a neutral amino acid (valine) to an acidic amino acid (glutamate) in mouse SDHC, is located within the functional ubiquinone-binding region of complex II. The cell lines expressing the same amount of mRNA in transgenic and endogenous wild-type alleles were named SDHC E69, and SDHC E69 cell lines were used as mev-1 cells. 41 Mev-1 cells accumulate cytoplasmic carbonyl protein and 8-oxoguanine at a faster rate than wild-type cells. In the first month after establishment, mev-1 cells show a loss of contact inhibition, and there are many apoptotic molecular like particles. The activity of caspase 3 is 1.3–1.8 times higher. When one-month mev-1 cells were injected under the epithelium of nude mice, they rapidly disappeared as compared with the wild type. These cells undergo apoptosis. Conversely, injecting the same number of three-month mev-1 cells resulted in the production of tumors. 39
The mutation of mev-1 of Caenorhabditis elegans leads to substitution of the 71st amino acid from glycine to glutamic acid (G71E). 65 The ability of complex II to participate in electron transmission is impaired. The mev-1 mutation is allergic to the ROS producing chemical methyl viologen, and the rate of accumulating fluorescent substances and protein carbonyl derivatives is significantly higher than that of wild type.66,67 The mev-1 mutation is very sensitive to the increase in oxygen concentration. With the increase of oxygen concentration from 1% to 60%, its life span is significantly shortened. 68 Compared with the control flies, the average life span of transgenic flies expressing dominant-negative form SdhCI71E was significantly reduced by 22%, and the amount of carbonyl protein was significantly increased, indicating that these flies induced high levels of oxidative stress in vivo. 69 The drosophila model can be used to study the aging process induced by excessive ROS.
A mev-1 transgenic mouse that contained the mutated SDHCV69E transgene is the mutation site located in the ubiquinone functional-binding region of complex II. The level of ROS in the mitochondria of the heart and muscle of this mouse increased, and the weight and exercise ability decreased. Mitochondrial structure abnormalities, especially in muscle, showed swelling and enlargement, leading to atrophy of muscle fibers. In addition, this mev-1 transgenic mouse showed a sterile phenotype. 70 The Tet-mev-1 conditional transgenic mouse line was established through the Tet-On/Off structure. 71 It has low fertility, is prone to spontaneous abortion and repeated abortion, and age-related female infertility, low birth weight and growth retardation, and accumulates cytoplasmic carbonyl protein and 8-oxguanine faster than wild-type mice.71 –73 Middle-aged Tet-mev-1 mice showed JNK/SAPK activation and Ca2+ overload, particularly in astrocytes. This led to decreasing levels of glial fibrillary acidic protein and S100β in the hippocampal area, and astrocyte deficiency occurred. 30 Tet-mev-1 mice exhibited accelerated corneal dysfunction with age, that is, delayed keratitis epithelialization, decreased endothelial cells, thickened Descemet’s membrane, and corneal stroma thinning. It can also cause dry eye syndrome due to lacrimal gland dysfunction.74 –76 In addition to being a mitochondrial-mediated oxidative stress model, this model can also be used to study the effects of oxidative stress on neurons or astrocytes and the pathogenesis of ROS in the eye.
Possible therapeutic targets
MCII is a pharmacologically useful target because drugs can relatively independently interfere with its two enzymatic activities: SDH activity in maintaining the TCA cycle and SQR activity in transporting electrons from the TCA cycle to the respiratory chain. 77 The natural compound gracillin disrupts MCII function by eliminating SDH activity without affecting SQR, with potential as an antitumor drug. 78 SDH inhibitors target SDH on the mitochondrial respiratory transport chain, cover ubiquinone sites, block the transmission of electrons from [3Fe-4S] to coenzyme Q, interfere with respiration, lead to ROS production, and eventually, cause cell apoptosis. Moreover, they are widely used as insecticides in fungal diseases worldwide.79,80 SDHC is lined at the QP site and contributes to formation of the QP site in MCII. 81 The QP site can be used as a target for cell death induction related to cancer therapy. 82 Mitochondrial complex II inhibitor, which specifically inhibits the ubiquinone-binding site of SDH, can enhance the apoptosis induced by cisplatin, a drug commonly used in cancer treatment. This discovery is helpful to solve the drug resistance of chemotherapy in cancer treatment and the serious side effects of patients. 83 Patients with SDHC epimutation paraganglioma may benefit from drugs causing hypomethylation and targeting hypoxia-inducible factors. 53 ASV has a dominant-negative effect on SDHC activity. Compared with the full-length isoform-expressing cells, SDHC Δ5 ASV expression significantly reduces SDH activity, providing a new therapeutic target, possibly by promoting the production of non-functional or dominant-negative types of SDHC ASV to limit the growth potential of tumor cells. 45 Further studies are required to identify specific inhibitors or activators of SDHC.
Conclusions
Oxidative stress results from oxygen consumption in aerobic respiration by an organism and is expressed as a persistent state of imbalance between the production of ROS and detoxification capacity of the endogenous antioxidant system. As an important subunit of SDH, SDHC participates in the TCA cycle and electron transfer and plays a key role in mitochondrial metabolism. When SDHC is inactivated, abundant ROS is produced, which is a key factor in the occurrence and development of many diseases, such as neurodegenerative diseases. SDHC transgenic animal model is a suitable model to study oxidative stress of respiratory chain injury. However, SDHC regulation in SDH and SQR activities requires further research. It can advance our understanding of the mechanism of MCII damage in oxidative stress and induction of apoptosis and, simultaneously, promote our understanding of related diseases.
Footnotes
Authors’ Contributions
All authors participated in the design, interpretation, and analysis of the data and review of the manuscript; QW wrote the article, YLZ, ML, NNZ collected and analyzed the literature, and JGY modified the article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (NSFC, No. 81860246, 82160517), Thousands of Young and Middle aged Backbone Teachers in Guangxi Colleges and Universities Training Plan, and Innovation Project of Guangxi Graduate Education.