
Abstract
Hyperexcitability is a major mechanism implicated in several neuropsychiatric disorders, such as organophosphate-induced status epilepticus (SE), primary epilepsy, stroke, spinal cord injury, traumatic brain injury, schizophrenia, and autism spectrum disorders. Underlying mechanisms are diverse, but a functional impairment and loss of GABAergic inhibitory neurons are common features in many of these disorders. While novel therapies abound to correct for the loss of GABAergic inhibitory neurons, it has been difficult at best to improve the activities of daily living for the majority of patients. Alpha-linolenic acid (ALA) is an essential omega-3 polyunsaturated fatty acid found in plants. ALA exerts pleiotropic effects in the brain that attenuate injury in chronic and acute brain disease models. However, the effect of ALA on GABAergic neurotransmission in hyperexcitable brain regions involved in neuropsychiatric disorders, such as the basolateral amygdala (BLA) and CA1 subfield of the hippocampus, is unknown. Administration of a single dose of ALA (1500 nmol/kg) subcutaneously increased the charge transfer of inhibitory postsynaptic potential currents mediated by GABAA receptors in pyramidal neurons by 52% in the BLA and by 92% in the CA1 compared to vehicle animals a day later. Similar results were obtained in pyramidal neurons from the BLA and CA1 when ALA was bath-applied in slices from naïve animals. Importantly, pretreatment with the high-affinity, selective TrkB inhibitor, k252, completely abolished the ALA-induced increase in GABAergic neurotransmission in the BLA and CA1, suggesting a brain-derived neurotrophic factor (BDNF)-mediated mechanism. Addition of mature BDNF (20 ng/mL) significantly increased GABAA receptor inhibitory activity in the BLA and CA1 pyramidal neurons similar to the results obtained with ALA. ALA may be an effective treatment for neuropsychiatric disorders where hyperexcitability is a major feature.
Keywords
Impact Statement
The work presented in this article shows that the nutraceutical alpha-linolenic acid (ALA) significantly increases GABAA receptor inhibitory activity in BLA and CA1 pyramidal neurons. A BDNF-TrkB-mediated mechanism is suggested based on (1) a similar increase in GABAA receptor inhibitory by BDNF in BLA and CA1 pyramidal neurons in slices from naïve young adult male Sprague-Dawley rats and (2) the high-affinity TrkB receptor antagonist, k252a, completely abolishes the ALA-induced increase in GABAA receptor inhibitory activity in BLA and CA1 pyramidal neurons. To our knowledge, this is the first report showing that ALA elicits an inhibitory activity in the brain. We propose that neuroprotection by ALA is mediated via a bidirectional effect by reducing excitation and enhancing inhibition. The enhanced GABAergic activity by ALA may restore the balance between excitation and inhibition in the central nervous system (CNS).
Introduction
Hyperexcitability has emerged as a major mechanism implicated in network circuitry dysfunction involved in seizures, cognitive impairment, and emotional disorders in a wide variety of neuropsychiatric disorders. Neurological disorders, such as traumatic brain injury,1 –3 stroke,4,5 epilepsy, 6 Alzheimer’s disease and Parkinson’s disease, 7 autoimmune disorders, 8 and paraneoplastic disorders, 9 have been shown to exhibit hyperexcitability in the CNS. Psychiatric disorders, such as posttraumatic stress disorder, addiction, and other acute stress syndromes, 10 autism spectrum disorders, 11 schizophrenia, 12 bipolar disorder, 13 and genetic disorders, such as Fragile X syndrome, 14 have all implicated hyperexcitability as a major pathophysiological mechanism. A common finding in these disorders is the dysfunction and loss of gamma-aminobutyric acid (GABA) inhibitory neurons.
Glutamate and GABA are the two major neurotransmitters in the nervous system. The vast majority of neurons are glutamatergic, whereas GABAergic inhibitory neurons represent about 15–20% of the total neuronal population.15,16 Despite the more numerous excitatory neurons relative to GABAergic inhibitory neurons, excitation and inhibition are balanced under physiological conditions in the CNS. Whereas excitatory neuronal function is involved in sensory, motor, and behavioral processes, excitatory–inhibitory balance dysfunction has been implicated in neuropsychiatric disorders. For example, excessive release and overactivation of glutamate receptors can result in neuronal cell death by domoic acid,17,18 the neurotoxin responsible for the neurologic consequences in humans after eating contaminated mussels. 19 Neurological findings include alterations in consciousness, short-term memory impairment, seizures, headache, and neurologic deficits among others. Neuropathological analysis showed neuronal cell death in the amygdala and hippocampus,20,21 two limbic brain regions involved in kainic-acid-induced neuronal cell death, and in temporal lobe epilepsy. 22 Palytoxin 23 and okadaic acid 24 are two additional marine toxins that can cause neuronal cell death in culture.
To maintain the excitatory–inhibitory balance in the CNS, GABAergic inhibitory neurons exhibit diverse morphological and physiological properties to mediate their effects on excitatory and other GABAergic inhibitory neurons. 25 GABAergic synaptic transmission is critical for temporal neural circuitry precision and synchronous oscillations of neuronal populations influencing the flow of excitation in brain regions. The activity of GABAergic inhibitory neurons is an important mechanism coordinating groupings of excitatory neuronal networks, either locally or remotely, facilitating the complex actions of the brain. 26 Thus, neuronal inhibition through hyperpolarization dynamically controls excitation in diverse neuronal circuitry arrangements.27,28
GABAergic inhibitory neurons are expressed at high levels in several brain regions, including the neocortex, hippocampus, and cerebellum. Impairment and loss of any part of this complex system can alter cellular function and behavior.29 –31 Chronic stress, for example, can alter GABAergic inhibitory neuronal function in the basolateral amygdala (BLA), resulting in hyperexcitability of outflow of principal excitatory neurons culminating in fear and anxiety.10,32
Reduced GABAA receptor-mediated inhibition has been suggested as a mechanism of hyperexcitability in patients with Fragile X syndrome. 14 Our group showed that hyperexcitability in the BLA after a mild traumatic brain injury is associated with an anxiety-like phenotype; 1 the anxiety-like phenotype, hyperexcitability, and GABAergic neuronal loss in the BLA were prevented by the nutraceutical ALA administered after the injury. 33 An anxiety-like phenotype develops 14 days after soman-induced SE and is associated with loss of GABAergic inhibitory neurons at day seven in the BLA. 34 Interestingly, disruption of the blood–brain barrier leading to extravasation of albumin results in the breakdown of protective extracellular matrix structures or perineuronal nets around specific GABAergic inhibitory neurons associated with hyperexcitability in a model of traumatic brain injury and in tissue from human epilepsy patients. The mechanism may involve transforming growth factor-beta (TGFβ) signaling in astrocytes. 5 In the autoimmune disorder, LG1 limbic encephalitis, auto-antibodies to leucine-rich glioma-inactivated protein (LGI1), reduced inhibition more than excitation resulting in an overall increase in network excitability in the hippocampus. 35 Neurological sequelae are critically dependent on the brain region(s) affected by hyperexcitability. However, GABAergic inhibitory neuronal impairment/loss plays a critical role in many of these neurologic and psychiatric disorders. Hyperexcitability can result in the overactivation of glutamate receptors leading to glutamate-mediated excitotoxic cell death. Indeed, glutamate-mediated excitotoxicity is thought to be a major mechanism in the neurological disorders discussed here.1 –7
ALA is an omega-3 polyunsaturated fatty acid found in plants. ALA reduces neuronal cell death by inhibiting glutamatergic neurotransmission via activating a relatively new class of potassium channels with two-pore-forming domains (TREK-1) that have potassium background channel function,36,37 are abundantly expressed in brain, 38 and are localized on presynaptic and postsynaptic neurons. 39 Synaptic terminal neuronal membranes are hyperpolarized by ALA activation of TREK-1 potassium channels decreasing glutamate release and hyperpolarizing postsynaptic neuronal membranes to leverage the blockade of the magnesium-associated channel of the N-methyl-D-aspartate (NMDA) receptor and inhibit the depolarization by the action of glutamate on other glutamate ionotropic receptors. 39 Subcutaneous (sc) or intravenous (iv) ALA administration after soman-induced SE significantly decreased neuronal degeneration at 24 h in the hippocampus, amygdala, piriform cortex, and cortex. Repeated ALA iv administration at 30 min, 3 days, and 7 days after soman (1) significantly increased animal survival at 21 days; (2) significantly reduced neuronal degeneration up to 21 days, 14 days after the last ALA injection suggesting that ALA exerted secondary effects; (3) significantly improved performance on the rotorod, and passive avoidance task at 15–26 days, and exerted an antidepressant-like effect in the forced swim test at day 15; (4) significantly increased mature brain-derived neurotrophic factor (BDNF) protein principally in hippocampal neurons, phospho-Akt, and phospho-mammalian target of rapamycin complex 1 (mTORC1) in the hippocampus; (5) significantly increased neurogenesis and the number of mature neurons at 31 days in the subgranular zone of the dentate gyrus; and (6) rapamycin, an inhibitor of mTORC1, blocked the ALA-induced neurogenesis and improved retention on the passive avoidance task suggesting that the mTOR activation was required for the ALA neurogenesis-induced improvement on performance tasks.40 –43 These results demonstrate the well-described pleiotropic properties of ALA that have also been shown in the normal brain. 44
Here, we show that ALA facilitates GABAergic neurotransmission in the BLA and CA1 subfield of the hippocampus via a TrkB-dependent mechanism to increase GABAA receptor inhibitory activity.
Materials and methods
Ethics statement
All animal experiments were conducted following the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Research Council) and were in accordance with the guidelines and approved by the Uniformed Services University of the Health Sciences Institutional Animal Care and Use Committees (IACUC). All efforts were made to minimize the number of animals used and any pain or distress associated with these experiments.
Animals and experimental groups
Experiments were performed using 6- to 10-week old male, Sprague-Dawley rats (Charles River, Wilmington, MA, USA). Rats were pair-housed on arrival and acclimated for three days. A total of 26 rats were used for the study. Animals were housed on an environmentally controlled room (20–23°C, ~44% humidity, 12-h light/12-h dark cycle [350–400 lux], lights on at 18:00), with food (Harlan Teklad Global Diet 2018, 18% protein rodent diet; Harlan Laboratories; Indianapolis, IN) and water available ad libitum. Overall, 10 animals were used to study the effects of ALA administered in vivo. Animals were initially divided into two study groups: ALA and vehicle (VEH). The animals were administered subcutaneously with a single dose of ALA (1500 nmol/kg) or VEH (0.05% ethanol). Animals were weighed prior to every injection to ensure that the proper ALA dose was injected subcutaneously; the identical volume of VEH was injected into VEH animals. A needle size ranging from 22 to 25 gauge was used and the injected volume was 0.25–0.35 mL. Animals from both groups were euthanized 24 h after injection, and brains were used in electrophysiological experiments.
Another 10 rats were used to study the effects of ALA applied to brain slices. The remainding six rats were used to test the effect of K252a applied before ALA and for the application of mature BDNF in brain slices. All 16 rats described were not injected with drugs prior to electrophysiological experiments.
Electrophysiological experiments
The procedures for obtaining the whole-cell recordings from the BLA and hippocampal CA1 region have been previously described.33,45 The protocol was the same for brain slices from ALA-injected animals and slices when ALA was bath-applied. The rats were anesthetized with isoflurane before decapitation. The coronal brain slices (400 µm thick) containing the amygdala and hippocampus were cut in ice-cold solution consisting in mM: 115 sucrose; 70 N-methyl-D-glucamine-gluconate (NMDG); 1 KCl; 2 CaCl2; 4 MgCl2; 1.25 NaH2PO4; 30 NaHCO3 with the use of a vibratome (Leica VT1200 S; Leica Microsystems, Buffalo Grove, IL, USA). The slices were transferred to a holding chamber at room temperature of about 23°C, in a bath solution containing in mM: 125 NaCl; 2.5 KCl; 1.25 NaH2PO4; 21 NaHCO3; 2 CaCl2; 1 MgCl2; and 25 D-glucose. The recording solution (artificial cerebrospinal fluid, ACSF) was the same as the holding bath solution. All the solutions were saturated with 95% O2/5% CO2 (carbogen) to achieve a pH near 7.4. The recording chamber (0.7 mL capacity) had continuously flowing ACSF (~8 mL/min) at 30–31°C. The osmolality of the ACSF was adjusted to 330 mOsm with D-glucose.
To visualize the neurons, we used a 40x water immersion objective equipped with a CCD-100 camera (Dage-MTI, Michigan City, IN, USA) under infrared light, using Nomarski optics of an upright microscope (Zeiss Axioskop 2, Thornwood, NY, USA). The recording electrodes had resistances of 3.5~4.5 mW when filled with the internal solution consisting in mM: 60 CsCH3SO3; 60 KCH3SO3; 5 KCl; 10 EGTA; 10 HEPES; 5 Mg-ATP; 0.3 Na3GTP (pH 7.2; osmolality was adjusted to 300 mOsm with potassium gluconate). Tight-seal (over 1 Giga Ohm) whole-cell recordings were obtained from the cell body of the principal neurons, distinguished from the interneurons by their larger size, pyramidal shape, and electrophysiological characteristics. Access resistance not exceeding 10 Mega Ohms was monitored during the recordings, and the cells were rejected if the resistance changed by more than 15% during the experiment. The currents were amplified and filtered (1 kHz) using the Axopatch 200B amplifier (Axon Instruments, Foster City, CA, USA) with a four-pole, low-pass Bessel filter, digitally sampled (up to 2 kHz) using the pClamp 10.7 software (Molecular Devices, Sunnyvale, CA, USA) and subsequently analyzed using Origin2019b software (OriginLab Corporation, Northampton, MA, USA).
GABAA receptors (GABAARs)-mediated spontaneous inhibitory postsynaptic potential currents (sIPSCs) were recorded in a voltage-clamp mode at holding potential (Vh) of + 30 mV in the presence of D-AP5 (50 μM); SCH50911 (10 μM); and LY341495 (3 μM). After a BLA or hippocampal CA1 pyramidal neuron was patch-clamped, the holding potential was switched from conventional − 58 to + 30 mV. The cell was left to equilibrate with the new Vh for about 4 min in drug-free bath solution (ACSF) and then another 4 min in antagonists-containing bath solution. Spontaneous IPSCs were recorded during 4 min after that. Charge transferred by sIPSCs was estimated for 20-s-long time window. Pressure application of GABA (400 µM) was performed with the help of the technique described previously. 46 Drugs used in this study were as follows: D-AP5, a competitive NMDA receptor antagonist, SCH50911, a GABAB receptor antagonist, bicuculline, a GABAA receptor antagonist, and all chemicals used for buffers were purchased from Sigma-Aldrich Chemical Co (St. Louis, MO). LY341495, a metabotropic glutamate group II/III receptor antagonist and K252a, a selective inhibitor of Trk receptors, were purchased from Tocris Bioscience (Ellisville, MO). BDNF was purchased from R&D Systems, Inc. (Minneapolis, MN).
ALA
ALA (Nu-Chek Prep Inc, Elysian, MN) was freshly prepared on the day of experimentation. ALA was dissolved in ethanol at a molar concentration and then diluted in NaCl 0.9% solution to reach a final concentration of 500 µM. The pH of the solution was adjusted to 7.0. VEH (0.05% ethanol) was prepared in an identical fashion and served as the appropriate control for ALA-injected animals and for in vitro experiments. ALA is known to undergo auto-oxidation. 47 Precautious included performing all manipulations and storage of ALA under nitrogen.
Statistical analysis
Statistical values are presented as mean ± standard error of the mean (SEM). Results from animals that received VEH were compared with the results of animals that received ALA using the independent t-test. Results from application of VEH or ALA in slices were compared using paired t-test. Results were considered statistically significant when the p-value was less than 0.05. Sample sizes (n) refer to the number of neurons.
Results
However, 24 h after injection of ALA or VEH, recordings from CA1 and BLA show an increase in the inhibitory activity mediated via GABAA Rs for animals that received ALA (Figures 1 and 2). To determine if this effect was due to the direct action of ALA in the brain, we recorded from CA1 and BLA pyramidal neurons from animals that did not receive any treatment.
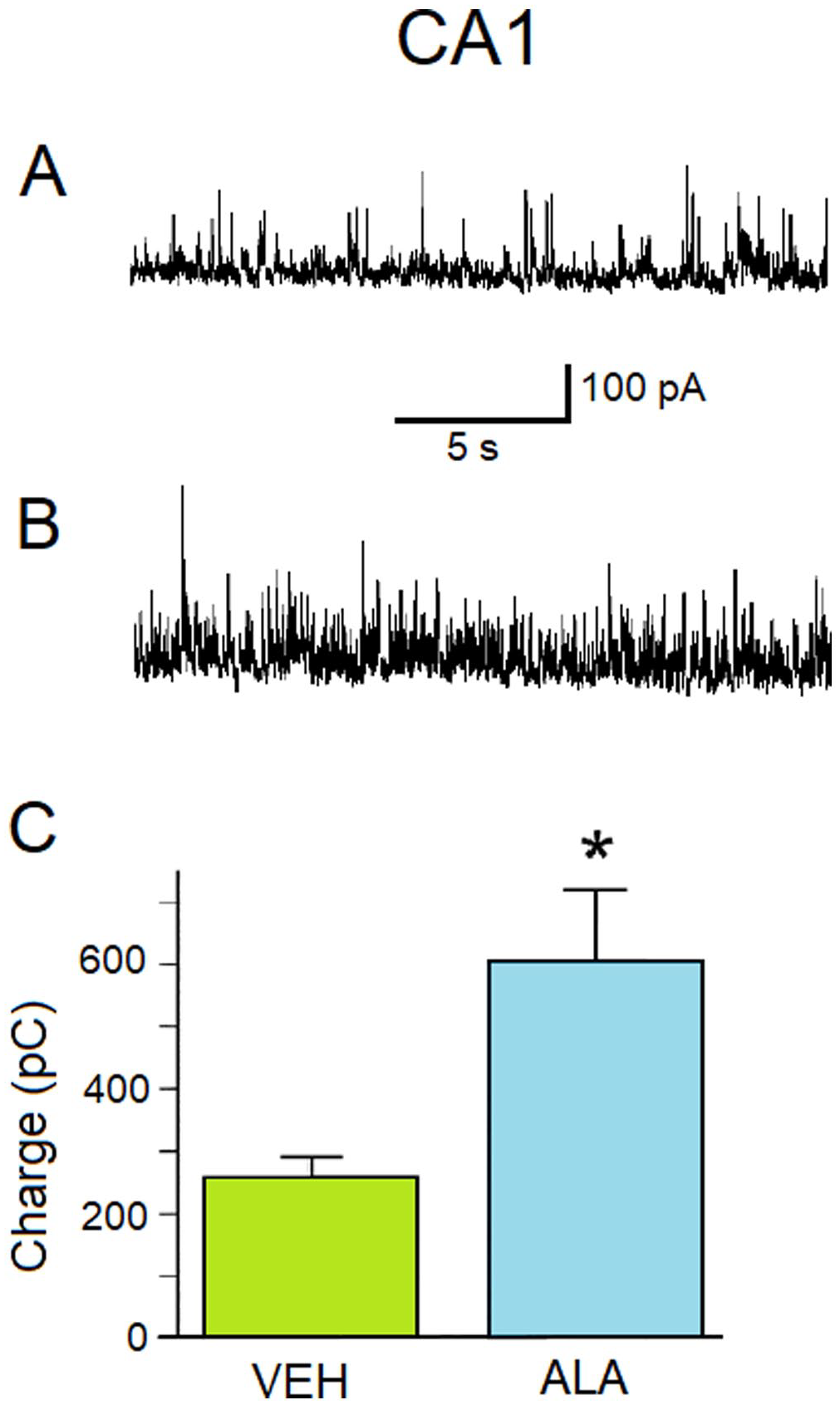
ALA increases GABAA receptor-mediated inhibitory activity in the CA1 subfield of the hippocampus. (A) Representative current traces of sIPSCs with a holding potential of + 30 mV recorded from the CA1 subfield of the hippocampus from 6- to10-week old male Sprague-Dawley rats injected subcutaneously (sc) with a single dose of VEH (0.05% ethanol) 24 h after injection. (B) Representative current traces of sIPSCs with a holding potential of + 3 mV recorded from the CA1 subfield of the hippocampus from 6- to 10-week old male Sprague-Dawley rats injected sc with a single dose of ALA (1500 nmol/kg) 24 h after injection. (C) The charge transferred by sIPSCs measured in pico-Coulombs (pC) (ordinate axis). Histograms showing the amount of charge transferred by sIPSCs during 20 s at Vh + 30 mV for different animals injected with either VEH or ALA (abscissa axis). VEH charge 260 ± 30 pC; n = 4. ALA histogram: charge 600 ± 100 pC; n = 5. The difference between histograms is statistically significant (two-sample t-test; *p = 0.0309).
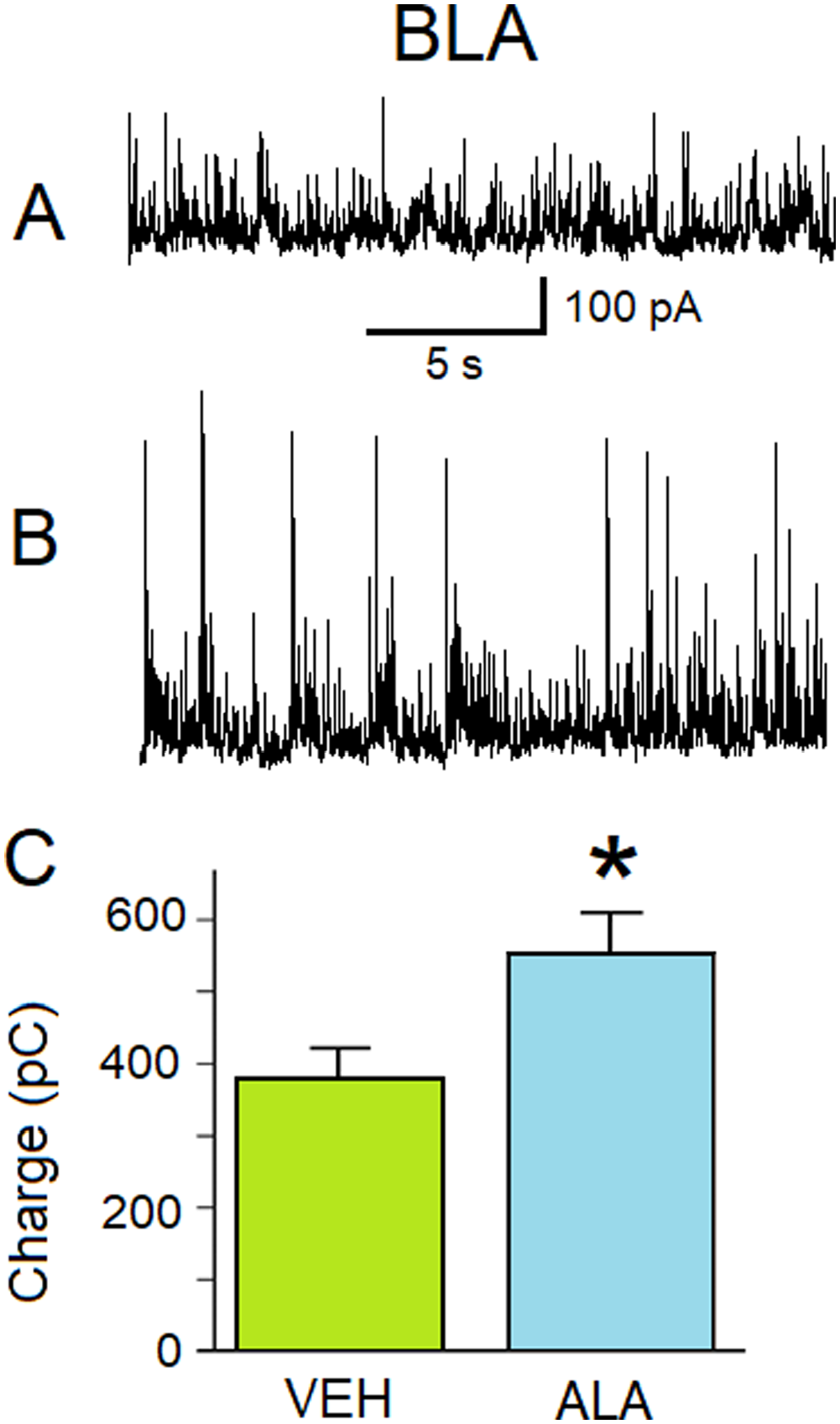
ALA increases GABAA receptor-mediated inhibitory activity in the BLA. (A) Representative current traces of sIPSCs at a holding potential of + 30 mV recorded from BLA principal neurons from 6- to 10-week old male Sprague-Dawley rats injected sc with a single dose of VEH (0.05% ethanol). (B) Representative current traces of sIPSCs at a holding potential of + 30 mV recorded from BLA principal neurons from 6- to 10-week old male Sprague-Dawley rats injected sc with a single dose of ALA (1500 nmol/kg) 24 h after injection. (C) The charge transferred by sIPSCs measured in pico-Coulombs (pC) (ordinate axis). The histograms illustrating the amount of charge transferred by sIPSCs during 20 s at Vh + 30 mV for different animals in different conditions (abscissa axis). “VEH” histogram: charge 380 ± 40 pC; n = 21. ALA histogram: charge 550 ± 60 pC; n = 19. The difference between columns is statistically significant (two-sample t-test; *p = 0.02793).
Recordings were made in solution containing D-AP5, SCH50911, and LY341495. At a holding potential of + 30 mV, all outward currents recorded from CA1 and BLA pyramidal neurons were mediated by GABAARs as proven by bath application of solution containing 20 µM bicuculline (Figure 3). IPSCs and burst IPSCs were blocked by 20 µM bicuculline (Figure 3[A] and [B]). Bath application of ALA (freshly bubbled with carbogen, about 8 mL/min) to CA1 pyramidal neurons lead to an increase in the inhibitory activity mediated via GABAARs (Figure 4). Bath application of ALA to BLA principal pyramidal neurons also lead to an increase in the inhibitory activity mediated via GABAARs (Figure 5). We tested whether the effect of ALA, that was bath-applied to principal BLA neurons, may be linked to the direct facilitation of GABAARs. We pressure-applied 200 ms pulses of 400 µM GABA to the patch-clamped BLA principal neurons while performing bath application of ALA (100 µM). The amplitude of GABA-evoked outward currents did not change while the facilitating effect of ALA bath application was observed (results not shown).
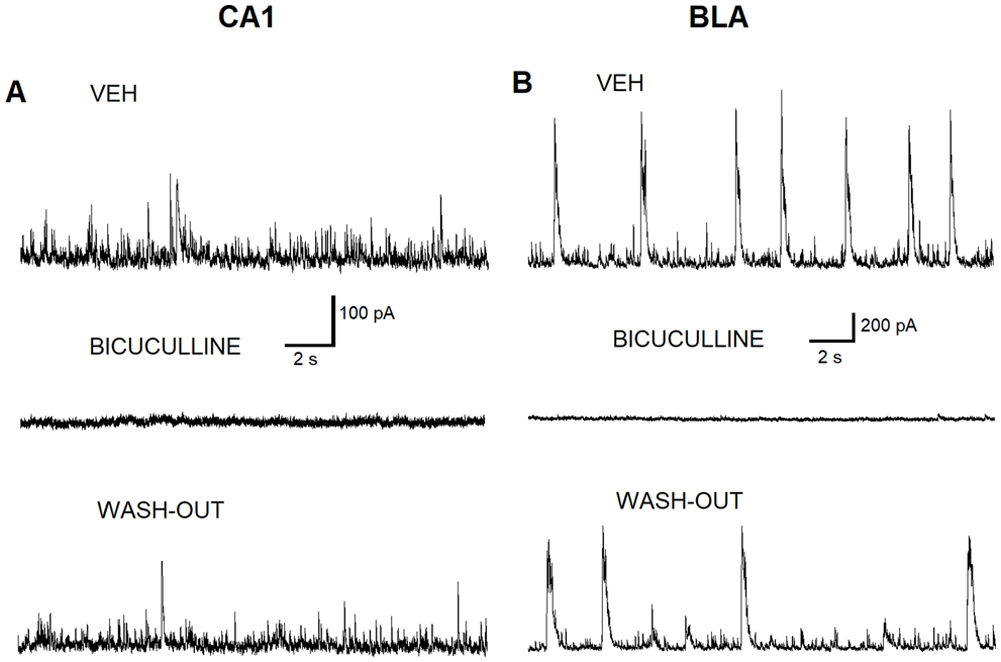
Bicuculline blocks the GABAAR inhibitory activity induced by ALA in CA1 and BLA pyramidal neurons. (A) Representative outward currents (sIPSCs) recorded from CA1 pyramidal neurons in the bath solution containing 0.05% ethanol (VEH) (upper panel). Addition of bicuculline (20 µM) blocks sIPSCs when bath-applied to BLA pyramidal neurons (Middle panel). Washout of the bicuculline from the bath restores sIPSCs (lower panel). (B) Representative outward currents (sIPSCs) recorded from BLA pyramidal neurons in the bath solution containing VEH (upper panel). Addition of bicuculline (20 µM) blocks sIPSCs when bath-applied to BLA pyramidal neurons (middle panel). Washout of the bicuculline from the bath restores sIPSCs (lower panel).
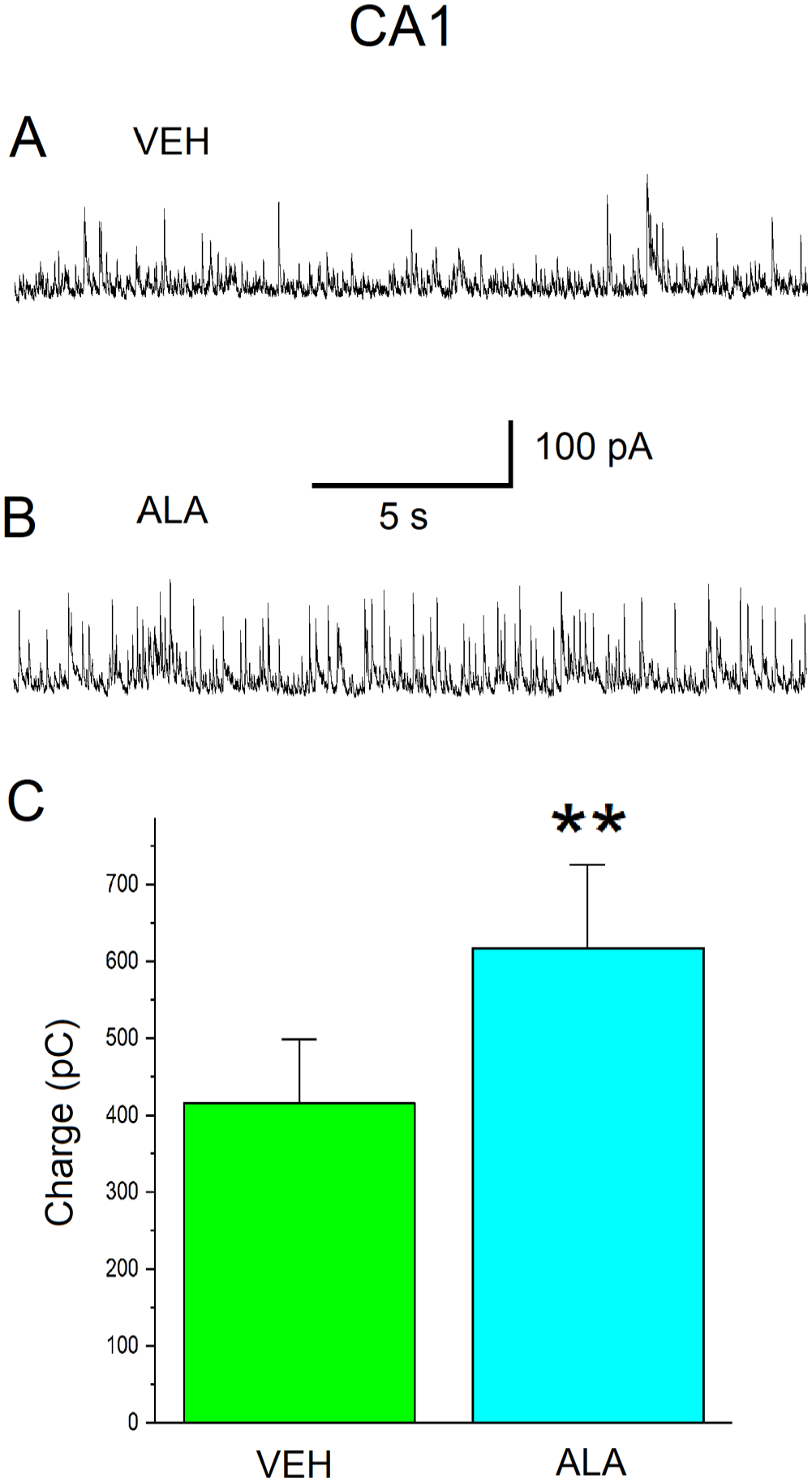
Bath-applied ALA increases GABAAR inhibitory activity in the CA1 subfield of the hippocampus in slices from naïve male Sprague-Dawley rats. (A) Representative current traces of sIPSCs at a holding potential of + 30 mV recorded from CA1 pyramidal neurons in the bath solution containing VEH (0.05% ethanol). (B) Representative current traces of sIPSCs at a holding potential of + 30 mV recorded from CA1 pyramidal neurons in the bath solution containing ALA (100 µM). (C) Charge transferred by sIPSCs is measured in pico-Coulombs (pC) (ordinate axis). Histograms illustrating the amount of charge transferred by sIPSCs during 20 s at Vh = + 30 mV for different cells in the slices (abscissa axis). VEH histogram: charge 420 ± 80 pC; n = 10. ALA histogram: charge 600 ± 100 pC; n = 10. The difference between the histograms is statistically significant (paired-sample t-test; **p = 0.00114).
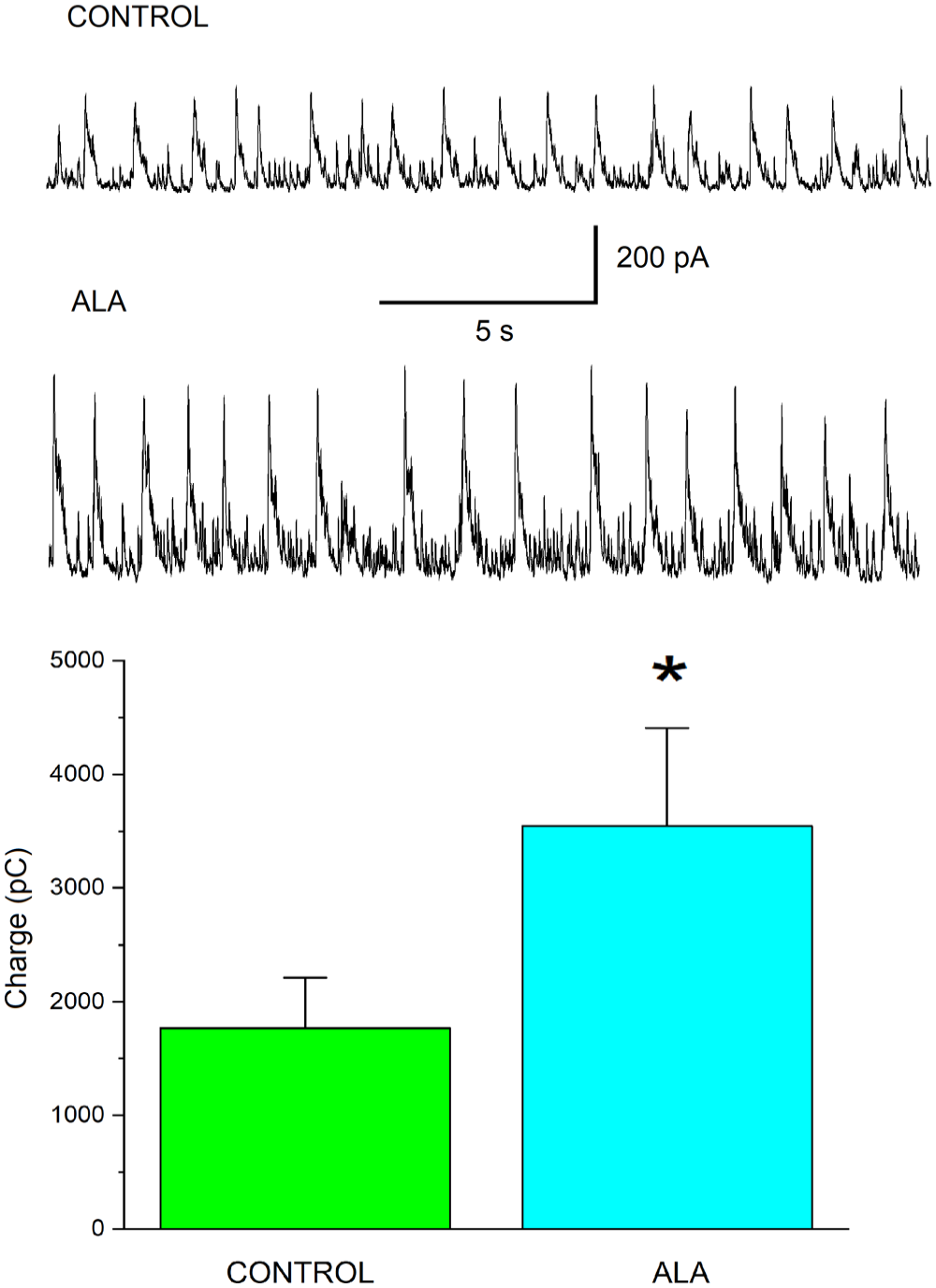
Bath-applied ALA increases GABAAR inhibitory activity in the BLA in slices from naïve male Sprague-Dawley rats. (A) Representative current traces of sIPSCs at a holding potential of + 30 mV recorded from BLA pyramidal neurons in the bath solution containing VEH (0.05% ethanol). (B) Representative current traces of sIPSCs at a holding potential of + 30 mV recorded from BLA pyramidal neurons in the bath solution containing ALA (100 µM). (C) Charge transferred by sIPSCs is measured in pico-Coulombs (pC) (ordinate axis). Histograms illustrating the amount of charge transferred by sIPSCs during 20 s at Vh = + 30 mV for different cells in the slices (abscissa axis). VEH histogram: charge 1800 ± 400 pC; n = 7. ALA histogram: charge 3600 ± 900 pC; n = 7. The difference between the histograms is statistically significant (paired-sample t-test; *p = 0.01858).
To test the hypothesis that the ALA effect may require the BDNF-TrkB pathway, we conducted the experiments on pyramidal neurons from BLA- and CA1-containing rat brain slices first without the TrkB antagonist K252a present in slice-holding chamber. Then similar experiments were conducted on BLA and CA1 pyramidal neurons from slices that were exposed to K252a (200 nM) for 1 h. Three CA1 neurons from the control batch of slices demonstrated an enhancement of GABAAR-mediated inhibitory activity by ALA (100 µM), whereas three CA1 neurons from the same holding chamber, but exposed to K252a, abolished the ALA-induced increase in GABAAR inhibitory activity (Figure 6[A] and [B]). Similar results were found for BLA neurons (Figure 7[A] and [B]). Direct bath application of mature BDNF (20 ng/mL) resulted in an increase in IPSCs both in CA1 (Figure 6[C]) and BLA (Figure 7[C]) pyramidal neurons similar to the initial increase in IPSC activity mediated via bath application of ALA (100 µM).
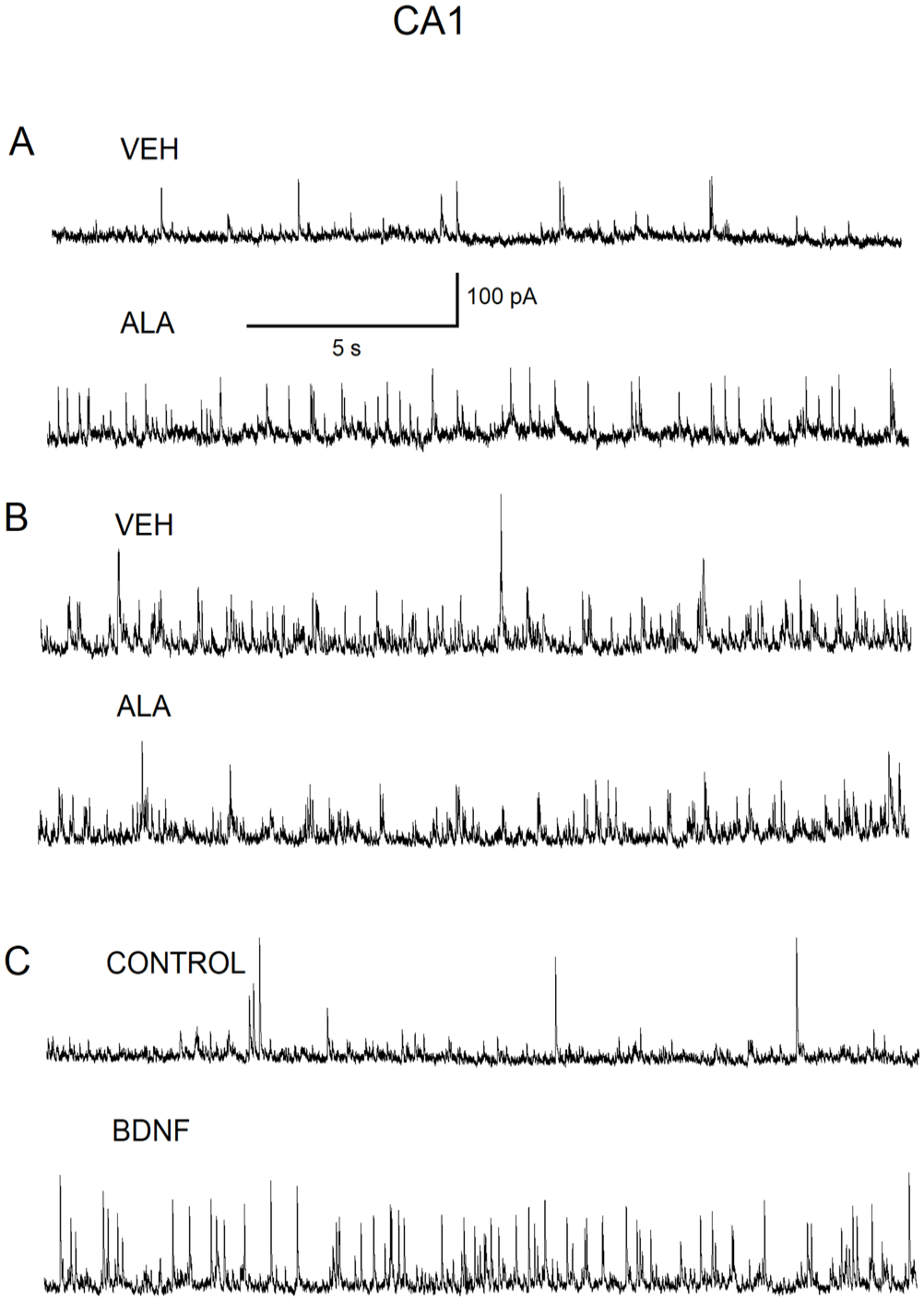
The high-affinity TrkB receptor antagonist, K252a, abolishes the ALA-induced increase in GABAAR inhibitory activity in CA1 pyramidal neurons. (A) Representative current traces of IPSCs recorded from CA1 pyramidal neurons from a slice that was held in the slice-containing chamber prior to the addition of K252a. Representative current IPSCs recorded from CA1 pyramidal neurons in the bath solution containing VEH (0.05% ethanol) (upper panel). Representative IPSCs recording from CA1 pyramidal neurons demonstrating the facilitation of inhibitory activity due to bath-applied ALA (100 µM) (lower panel). (B) Representative IPSCs recorded from CA1 pyramidal neurons from a slice that was held in slice-containing chamber for 1 h after the addition of K252a (200 nM) to the slice-containing chamber. IPSCs recorded in VEH (0.05%)-containing solution (upper panel). Lower recording: IPSCs recorded from CA1 pyramidal neurons after the addition of bath-applied ALA (100 µM) demonstrating no effect on recorded IPSCs (lower panel). (C) Representative IPSCs recorded from CA1 pyramidal neurons in control solution (upper panel). IPSCs recorded from CA1 pyramidal neurons after bath-applied mature BDNF (20 ng/mL) demonstrating the facilitation of GABAAR inhibitory activity due to bath-applied mature BDNF (20 ng/mL) (lower panel).
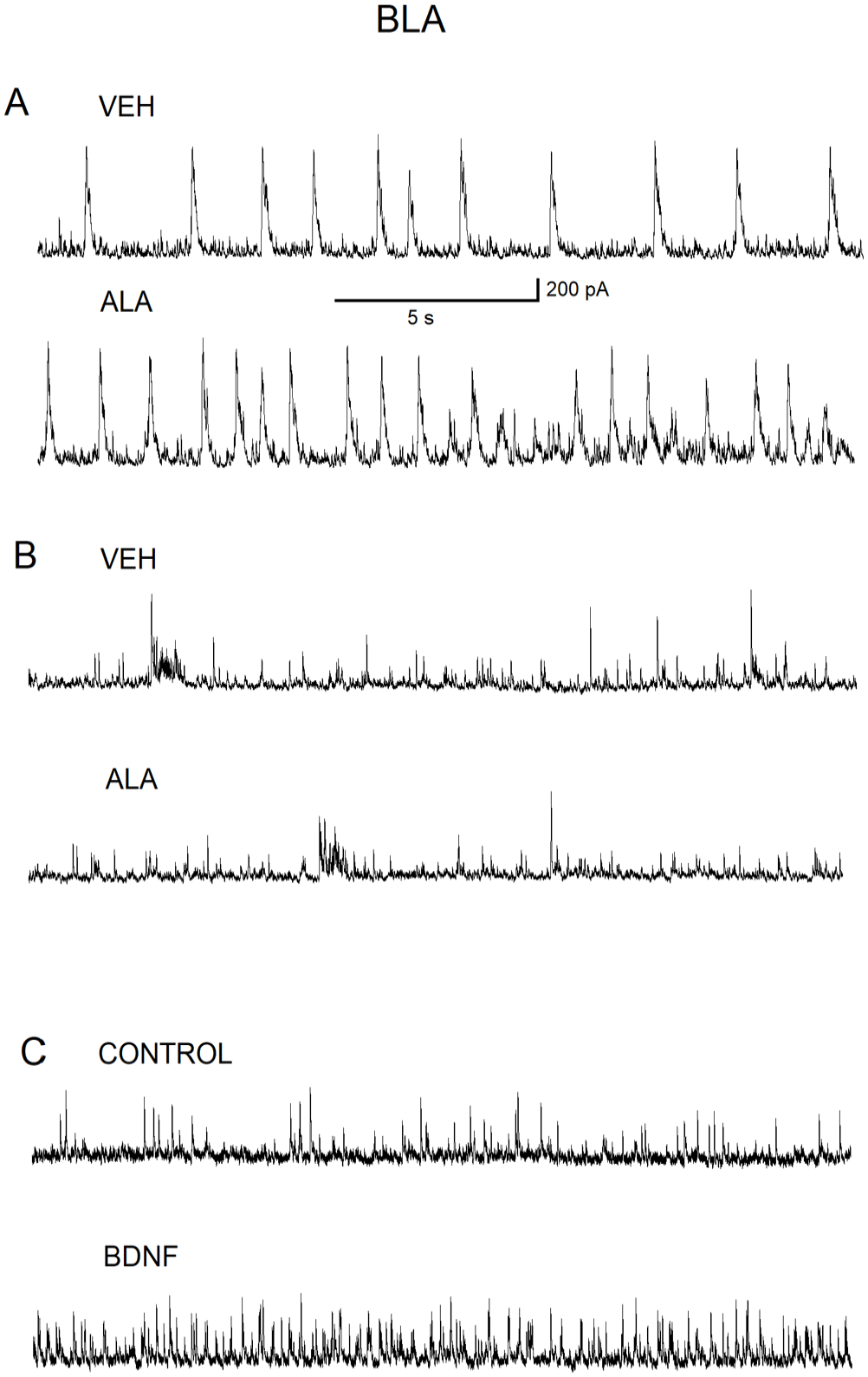
The high-affinity TrkB receptor antagonist, K252a, abolishes the ALA-induced increase in GABAAR inhibitory activity in BLA pyramidal neurons. (A) Representative current traces of IPSCs recorded from BLA pyramidal neurons from a slice that was held in the slice-containing chamber prior to the addition of K252a. Representative current IPSCs recorded from BLA pyramidal neurons in the bath solution containing VEH (0.05% ethanol) (upper panel). Representative IPSCs recording from BLA pyramidal neurons demonstrating the facilitation of inhibitory activity due to bath-applied ALA (100 µM) (lower panel). (B) Representative IPSCs recorded from BLA pyramidal neurons from a slice that was held in slice-containing chamber for 1 h after the addition of K252a (200 nM) to the slice-containing chamber. IPSCs recorded in VEH (0.05%)-containing solution (upper panel). Lower recording: IPSCs recorded from BLA pyramidal neurons after addition of bath-applied ALA (100 µM) demonstrating no effect on recorded IPSCs (lower panel). (C) Representative IPSCs recorded from BLA pyramidal neurons in control solution (upper panel). IPSCs recorded from BLA pyramidal neurons after bath-applied mature BDNF (20 ng/mL) demonstrating the facilitation of GABAAR inhibitory activity due to bath-applied mature BDNF (20 ng/mL) (lower panel).
Discussion
In this study, we show for the first time that a single subcutaneous injection of ALA increases GABAAR-mediated inhibitory activity in the BLA and CA1 pyramidal neurons 24 h after injection in young adult Sprague-Dawley male rats. These results propelled us to study this effect further by applying ALA (100 µM) to slices from young adult male rats. ALA increased GABAAR inhibitory activity as demonstrated by the increase in the charge transferred by outward GABAergic currents in voltage-clamp mode. The effect of the facilitation of inhibitory activity by ALA observed both in BLA and hippocampal CA1 pyramidal neurons was transient. The effect was reversible, however, the washout time was significant. The effect of ALA of much lower intensity could be observed only after more than 15- to 20-min washout of ALA.
To determine whether ALA-facilitated GABAAR-mediated inhibitory activity evoked a direct effect on postsynaptic principal neurons, pressure-applied 200 ms pulses of 400 µM GABA were applied to the patch-clamped BLA principal neurons while performing bath application of ALA (100 µM). The amplitude of GABA-evoked outward currents did not change while the facilitating effect of ALA bath application was observed. These results indicate that ALA does not modulate postsynaptic GABAARs to enhance inhibitory activity.
Low-level activation of NMDA receptors increases the release of mature BDNF that in turn binds to and activates its cognate receptor, TrkB, in an autocrine mechanism to establish a neuroprotective state against NMDA receptor-mediated excitotoxicity in cultured cerebellar granule cells 48 and in hippocampal neurons. 49 During development, GABAA receptors are excitatory and activation of these receptors is in parallel with the activated BDNF-TrkB signaling pathway to control proliferation, maturation, synapse formation, and plastic responses.
Activation of excitatory GABAAR increases the cell surface expression of GABAAR and increases the release of mature BDNF which in turn activates TrkB receptors to increase the membrane expression of GABAAR. 50 After development, GABAARs switch from excitatory to serving as the major inhibitory neurotransmitter in the CNS. Nevertheless, we hypothesize that ALA results in the release of mature BDNF from either presynaptic neurons 51 or astroglial cells 52 that in turn activates TrkB receptors in either an autocrine or paracrine fashion to increase GABAAR inhibitory activity in the BLA and CA1 subfield of the hippocampus. Preincubation with the high-affinity TrkB inhibitor, K252a, abolishes the facilitation of GABAAR inhibitory activity in the BLA and CA1 subfield of the hippocampus. To provide supportive evidence for the ALA-induced increase in GABAAR inhibitory activity, we added mature BDNF (20 ng/mL) to the bath. A rapid increase in IPSC activity was observed after bath application of mature BDNF in the BLA and CA1 subfield of the hippocampus that was similar to the initial increase in IPSC activity mediated via bath application of ALA (100 µM).
The human bdnf gene is more than 70 kb and consists of 11 exons.53,54 The synthesized precursor protein is cleaved into pro-BDNF and pro-BDNF is cleaved into mature BDNF. 55 A major function of pro-BDNF is to mediate cell death, 56 whereas mature BDNF promotes neuronal survival. 57 We propose that ALA mediates the release of mature BDNF but the exact mechanism is unknown. In support of our findings, one study showed an increase in GABAAR currents by exposure to mature BDNF that was abolished by K252a in epileptic tissue from adult human brain. 58 Furthermore, it is known that astrocytes affect neuronal function. For example, IL-1β increases BDNF mRNA expression in hypothalamic neurons in culture but has the opposite response in the presence of astrocytes and appeared to be due to the release of prostaglandin E2 (PGE2) from astrocytes; addition of PGE2 to the culture medium reproduced the effect of IL-1β increase and decrease in BDNF mRNA in the enriched neuronal cultures and the co-cultures of astrocytes and neurons, respectively. 59 Interestingly, other omega-3 polyunsaturated fatty acids, such as docosahexaenoic acid (DHA), have been shown to increase mature BDNF protein levels in hippocampus by activating the GPR40 receptor. This receptor is expressed in the CNS and is activated by unsaturated fatty acids; 60 ALA like DHA influxes calcium and increases the phosphorylation (activation) of extracellular-regulated kinases 1 and 2. 61 It is possible that ALA binds to and activates GPR40 which in turn influxes calcium and the release of BDNF. Alternatively, it is possible that ALA elicits epigenetic changes that increase the levels of BDNF mRNA but longer exposure times are likely required. 62 However, it has been shown that BDNF reduces IPSCs in cerebellar granule cells and hippocampal neurons in culture.63,64 These conflicting results may be explained by the fact that our studies were performed in brain slices where network circuitry remains intact, whereas the above studies were performed in enriched isolated neuronal cultures in vitro.
Taken together, our findings show that ALA increases GABAAR inhibitory activity in BLA and CA1 pyramidal neurons in vivo and in brain slices from normal young adult male Sprague-Dawley rats. To our knowledge, this is the first report showing that ALA elicits an inhibitor activity. We further show that the ALA-induced increase in GABAAR inhibitory activity is replicated by the bath application of mature BDNF. The addition of the high-affinity TrkB inhibitor, k252a, abolishes the ALA-induced increase in GABAAR inhibitory activity in BLA and CA1 pyramidal neurons suggesting that ALA increases the release of mature BDNF that in turn increases GABAAR inhibitory activity by as yet an unknown mechanism.
There are potential broad implications to our results. It has been reported that the activation of background potassium channels leading to hyperpolarization of presynaptic and postsynaptic neurons that reduces the release of glutamate presynaptically and reduces the relief of the magnesium block of NMDA receptors postsynaptically is a major neuroprotective mechanism of ALA. Our results suggest that ALA may have opposing effects on the glutamatergic and GABAergic systems under excitotoxic conditions, that is, stroke, SE. ALA may elicit its neuroprotective effect(s) by reducing the excitotoxic effect of glutamate and enhancing the inhibitory GABAergic system on neurons at least in part through a BDNF-TrkB-mediated pathway. While novel therapies that reduce hyperexcitability in brain regions thought to play a major role in the pathophysiology in some neuropsychiatric disorders are likely to become available in the near future, 65 modulators of excitability, such as ALA, in an oral form may be a pragmatic and viable treatment strategy.
Footnotes
Authors’ Note
The opinions and assertions expressed herein are those of the author(s) and do not reflect the official policy or position of the Uniformed Services University of the Health Sciences or the Department of Defense.
Authors’ Contributions
All authors participated in the design, interpretation of the studies, analysis of the data, and review of the article. Electrophysiology experiments were conducted by VIP; animal experiments were conducted by HP; figures were prepared and analyzed by VIP and THF; article was written by AMM; and all authors edited the article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health Countermeasures against chemical threats (CounterACT) (grant nos.: 5U01NS102135-02 [MFMB] and 1U01NS123252-01A [MFMB]).