
Abstract
Src homology 2 domain–containing protein tyrosine phosphatase 1 (SHP-1) has a well-established role in myocardial infarction, yet its involvement in atrial fibrosis and atrial fibrillation (AF) has not been elucidated. As cardiac arrhythmias caused by AF are a major global health concern, we investigated whether SHP-1 modulates AF development. The degree of atrial fibrosis was examined using Masson’s trichrome staining, and SHP-1 expression in the human atrium was assessed using quantitative polymerase chain reaction (qPCR), immunohistochemistry (IHC), and western blotting (WB). We also examined SHP-1 expression in cardiac tissue from an AF mouse model, as well as in angiotensin II (Ang II)-treated mouse atrial myocytes and fibroblasts. We found that SHP-1 expression was reduced with the aggravation of atrial fibrosis in clinical samples of patients with AF. SHP-1 was also downregulated in the heart tissue of AF mice and Ang II-treated myocytes and fibroblasts, compared with that in the control groups. Next, we demonstrated that SHP-1 overexpression alleviated AF severity in mice by injecting a lentiviral vector into the pericardial space. In Ang II-treated myocytes and fibroblasts, we observed excessive extracellular matrix (ECM) deposition, reactive oxygen species (ROS) generation, and transforming growth factor beta 1 (TGF-β1)/mothers against decapentaplegic homolog 2 (SMAD2) pathway activation, all of which were counteracted by the overexpression of SHP-1. Our WB data showed that STAT3 activation was inversely correlated with SHP-1 expression in samples from patients with AF, AF mice, and Ang II-treated cells. Furthermore, administration of colivelin, a STAT3 agonist, in SHP-1-overexpressing, Ang II-treated myocytes and fibroblasts resulted in higher levels of ECM deposition, ROS generation, and TGF-β1/SMAD2 activation. These findings indicate that SHP-1 regulates AF fibrosis progression by modulating STAT3 activation and is thus a potential treatment target for atrial fibrosis and AF.
Impact Statement
Src homology 2 domain–containing protein tyrosine phosphatase 1 (SHP-1) expression is inversely related to atrial fibrillation (AF) severity, and its overexpression represses AF via dephosphorylating STAT3, leading to reduced atrial fibrosis, reactive oxygen species generation, and excessive extracellular matrix deposition. Therefore, overexpression of SHP-1 may improve AF, highlighting its potential as a promising target for AF treatment.
Introduction
Cardiac arrhythmias are highly prevalent and are associated with a high mortality rate worldwide. However, the molecular mechanisms associated with arrhythmia occurrence, propagation, and maintenance remain unclear. 1 Atrial fibrillation (AF) is generally associated with atrial fibrosis,2,3 which is often caused by extracellular matrix (ECM) protein precipitation and non-myocyte growth.4,5
Src homology region 2 domain–containing phosphatase-1 (SHP-1) is a non-transmembrane protein tyrosine phosphatase typically generated in hematopoietic progenitor cells. 6 SHP-1 negatively modulates signal transduction by phosphorylating receptors and is essential for cell death and/or repression of survival. 7 SHP-1 can inhibit the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway by dephosphorylating and inactivating STAT3, 8 a key regulator of cell apoptosis and inflammation signaling pathways. 9 STAT3 inactivation by SHP-1 is associated with knockdown of Y-box binding protein 1, which suppresses myocardial cell death mediated by hypoxia/reoxygenation. SHP-1-elicited STAT3 inactivation in hypoxia/reoxygenation in mice was also found to be closely related to the amelioration of acute myocardial infarction, a decreased infarct area, the oxidative stress response, and myocardial cell death. 10 Furthermore, Liu et al. showed that SHP-1 plays a myocardial protective role in hypoxia/reoxygenation-exposed cells and ischemia-reperfusion rats by mediating the STAT3 signaling axis. Furthermore, functional gain studies have shown that SHP-1 overexpression exerts antiapoptotic and anti-inflammatory effects. 11 Although the role of SHP-1 in myocardial infarction has been well-documented, its involvement in atrial fibrosis and AF has not been widely investigated.
Atrial fibroblasts are the main component of non-myocytes and a major ECM protein source in the atrium. 5 The repression of fibrosis in myocytes and atrial fibroblasts shows potential for AF treatment. In this study, we investigated the hypothesis that SHP-1 is involved in atrial fibrosis by modulating STAT3 activation in an AF cell model established by treating both atrial fibroblasts and myocytes with angiotensin II (Ang II). We investigated the mechanism underlying SHP-1 dysregulation in atrial fibrosis and AF to provide insight into approaches for preventing and treating AF.
Materials and methods
Ethics statement
All patients provided oral and written informed consent, and the study was strictly performed according to the principles of the Declaration of Helsinki. Animal experiments conformed to the guidelines for animal care and use. This study was approved by the Ethics Committee of Henan Provincial People’s Hospital, Fuwai Central China Cardiovascular Hospital.
Patients
Twenty patients who underwent heart surgery were recruited from October 2019 to May 2022 at Henan Provincial People’s Hospital and Fuwai Central China Cardiovascular Hospital. We included 10 patients with AF and 10 patients with sinus rhythm (SR) who met the following criteria: (1) heart function above class I–III evidence from the New York Heart Association; (2) without hypertension, hyperthyroidism, cardiomyopathy, diabetes mellitus, coronary heart disease, and malignancies; (3) <70 years old; and (4) discontinuation of angiotensin receptor blockers or angiotensin-converting enzyme inhibitors for no less than half a year prior to surgery.
Fresh right atrial appendage tissues were preserved using two protocols. One portion was fixed in paraformaldehyde (4%) and embedded in paraffin for immunohistochemistry (IHC) and histological staining, and the other portion was frozen in liquid N2 and stored at −80°C for quantitative polymerase chain reaction (qPCR) and western blotting (WB).
Histological analysis
The right atrial appendage tissue samples were fixed using 10% formalin, paraffin embedded, and cut into 5-µM-thick sections. The sections were then subjected to Masson’s trichrome staining using a modified Masson’s trichrome stain kit (Solarbio Science & Technology Co., Ltd., Beijing, China) according to the manufacturer’s instructions. Briefly, the sections were stained in Harris hematoxylin for 5 min, in Ponceau-acid fuchsin staining solution for 10 min, and in Aniline Blue solution for 5 min at room temperature. Images were obtained using an Olympus IX73 microscope (Olympus Corporation, Tokyo, Japan). ImageJ software for Windows 1.52v (National Institutes of Health [NIH], Bethesda, MD, United States of America) was used to quantify the Masson’s trichrome positive staining area.
AF mouse model
An AF mouse model induced using Ang II was established after lentiviral vector (LV) injection in the heart as previously described. 12 C57BL/6 mice (male, 8 weeks old) were provided by Charles River (Beijing, China). After anesthesia using pentobarbital sodium (40 mg/kg), an incision was made in the chest cavity, the pericardial space was injected with LV (10 μL) containing negative control (LV-NC) or SHP-1 (LV-SHP-1) through 36-gauge needles, and an incision was made in the pericardium. Then, an osmotic minipump (Alzer Pumps Model 1002; ALZET, Cupertino, CA, The United States) containing 200 μL Ang II (0.25 mg/mL in phosphate buffer saline [PBS], 2 mg/kg day) or PBS was implanted into mice for 14 days. 13 The mice were randomly classified into four groups: sham (minipump with PBS, n = 8), Ang II (minipump with Ang II, n = 8), Ang II + LV-NC (LV-NC injection + minipump with Ang II, n = 8), and Ang II + LV-SHP-1 (LV-SHP-1 injection + minipump with Ang II, n = 8). Fourteen days after surgery, eight mice from each group were sacrificed. The blood and left atrium were harvested for subsequent experiments.
Electrophysiological analysis
Hearts were collected from mice and perfused with Tyrode’s solution (5.4 mM KCl, 1.8 mM CaCl2, 140 mM NaCl, 2 mM MgCl2, 10 mM glucose, and 10 mM HEPES, pH = 7.4) at 37°C for 10 min after careful removal of adjacent tissues. A silver bipolar electrode was placed on the appendages of two atriums, and bipolar cardiac rhythms were recorded using a bio-amplifier system (PowerLab 8/30; ADInstruments, Bella Vista, NSW, Australia). We measured the effective refractory period (ERP) of the left atrium through programmed atrial electrical stimulation (S2) with eight regularly paced beats on cycle lengths of 90, 120, and 150 ms. We examined the interatrial conduction time (IACT) with an interelectrode distance between right and left atrium of 10 mm and determined the incidence of AF and AF duration time using an S3 extra-stimulus pacing method as previously described. 14
Isolation and cultivation of mouse myocytes and fibroblasts
The mouse hearts were harvested 1 week after birth, rinsed with prechilled PBS, and the upper sections were cut into pieces and digested with collagenase II (3 mL, 0.1%). Dulbecco’s Modified Eagle Medium (DMEM) containing fetal bovine serum (10%) was added to the supernatant to stop digestion by collagenase II; this step was performed five times. The cells were centrifuged at 1000 r/min for 3 min and cultivated in DMEM containing fetal bovine serum (10%) and bromodeoxyuridine (0.1 mM), and non-adherent cardiomyocytes were cleared. These steps led to the production of cultures with approximately 90% myocytes, which were validated by positive immunocytochemical staining using cardiac troponin T.
For fibroblast isolation, the mouse atrium was isolated and cut into small pieces, and then uniformly dispersed in a petri dish (6 cm) for 15 min at 37°C. The attached pieces were cultivated in DMEM, which were comprised of nutrient mixture F-12, fetal bovine serum (10%), and penicillin/streptomycin (1%), at 37°C in a 5% CO2 atmosphere. Cardiac fibroblasts from the second and third passages were further analyzed.
AF mouse cell model
Ang II can induce the transformation of mouse fibroblasts and atrial myocytes into myofibroblasts. 15 To investigate the effects of SHP-1 on Ang II-induced fibrosis, pCMV-myc-SHP-1 cells were established by transient transfection as follows: mouse atrial myocytes and fibroblasts were inoculated into six-well plates. When the confluence reached 60–80%, the cells were transfected with pCMV-myc-SHP-1 or pCMV-myc-NC using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, Massachusetts, USA) according to the manufacturer’s instructions. The transfected cells were cultivated for 1 day prior to treatment with Ang II (10 nM, stock concentration: 8 mM), 16 harvested, and used for qPCR, WB, and other analyses. For colivelin treatment, the cells were incubated with 5 μM colivelin. 17
Histological analysis
Masson’s trichrome was applied to stain the atrial appendage sections (4 μm) for fibrosis evaluation. The tissues were subjected to hematoxylin staining at 37°C for 5 min, and the proportion of the atrial area showing blue staining was determined to quantify the atrial collagen content.
IHC
Atrial appendage sections (4 μm) were subjected to deparaffinization and rehydration. Antigen retrieval was performed by heating the sections in sodium citrate (10%). Endogenous peroxidase activity was exposed to H2O2 (3%), blocking for 20 min at 37°C. The sections were incubated with primary antibodies at 48°C overnight, followed by incubation with secondary antibodies and detection with diaminobenzidine. SHP-1 (1:200; Abcam, Cambridge, United Kingdom) was used as the primary antibody. The stained slides were photographed (×200) using a BX51 microscope (Olympus, Tokyo, Japan), and positive staining was analyzed using Image-Pro Plus 6.0 software (Media Cybernetics, Rockville, MD, The United States).
qPCR
Total RNA was isolated using TRIzol® according to the manufacturer’s protocol. Prime Script Real-time Master Mix (TakaraBio, Kusatsu, Shiga Prefecture, Japan) was used to transcribe complementary DNA (cDNA). SHP-1 expression was determined using quantitative reverse transcription qPCR. The thermocycling procedure was as follows: predenaturation (95°C for 60 s), denaturation (95°C for 15 s), annealing (60°C for 40 s), and extension (72°C for 15 s) for a total of 40 cycles. Gene expression was determined using the 2–ΔΔCq method. GAPDH (Glycerinaldehyd-3-phosphat-Dehydrogenase) served as the internal reference.
WB
Total protein was isolated using a radioimmunoprecipitation assay lysis buffer and quantified using a Pierce™ BCA Protein Assay Kit (23225; Thermo Fisher Scientific, Waltham, MA, The United States). Proteins (20 µg total) were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (10%). The proteins were transferred from the gels to polyvinylidene fluoride membranes, which were blocked with fat-free milk (5%) for 2 h at RT and then incubated with primary antibodies overnight at 4°C. After rinsing three times with PBS, the membranes were incubated with an antibody labeled with a secondary polyclonal peroxidase (goat anti rabbit immunoglobulin G [IgG]; 1:4000; Abcam, Cambridge, The United Kingdom; cat. no. ab205718) for 2 h and observed using enhanced chemiluminescence. Beta-actin served as the loading control. ImageJ d1.47 software (NIH, Bethesda, MD, United States of America) was used to quantify the bands.
ROS detection
Reactive oxygen species (ROS) levels were examined as previously described. 18 Briefly, the cells were harvested, and the density was maintained at 106 cells/mL. CM-H2DCFDA (10 μM) was added to cells, followed by incubation for 1 h and three washes in PBS. Finally, ROS production in the cells was quantified using flow cytometry.
Data analysis
A two-sided t-test was used to analyze differences between two groups. One-way analysis of variance with Tukey’s multiple-comparisons test and two-way analysis of variance were also used to investigate differences between two groups and differences between two groups over time, respectively, using GraphPad Prism 7.0 software (GraphPad, Inc., La Jolla, CA, United States of America). The Shapiro–Wilk’s test was performed to test for Gaussian distribution of patient data. The results are presented as the mean ± standard error of the mean. Statistical significance is indicated as follows: *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
Clinical results
The statistics associated with the baseline clinical results of the patients are shown in Table 1. Electrocardiography and echocardiography indicated increased left and right atrial diameters in patients with AF compared with those of patients with SR. No significant differences in age, sex, left ventricular end-diastolic diameter, or ejection fraction were found between groups.
Clinical data of patients.

SR: sinus rhythm; AF: atrial fibrillation; M: male; F: female; LVEF: left ventricular ejection fraction; LVEDD: left ventricular end-diastolic dimension; LAD: left atrial dimension; RAD: right atrial dimension.
Compared with SR group, *P < 0.05.
SHP-1 downregulation in samples from patients with AF
Following Masson’s trichrome staining, the presence of blue collagen fibers, which were predominantly present in the myocardial interstitium, was used for the evaluation of atrial fibrosis. Both the degree of atrial fibrosis (Figure 1[A]) and proportion of fibrosis (Figure 1[B]) were dramatically higher in patients with AF than in patients with SR. A Shapiro–Wilk’s test was performed to test whether the data from 10 patients were Gaussian distributed. The W value was 0.913 (close to 1) and the P value was 0.632 (>0.05), suggesting that our data were Gaussian distributed. WB revealed that α-smooth muscle actin (SMA) and collagen I expression levels were remarkably higher in the AF group than in the SR group (Figure 1[C]). These data indicate a high level of fibrosis in the myocardial tissue of patients with AF. Next, qPCR, IHC, and WB were performed to detect SHP-1 expression in the myocardial tissue. The qPCR data showed that SHP-1 mRNA levels were lower in the AF atrium than in the SR atrium (Figure 1[D]). In the Shapiro–Wilk’s test, the W value was 0.966 (close to 1) and the P value was 0.688 (>0.05), suggesting that our data were Gaussian distributed. Immunostaining of the atrium revealed low SHP-1 expression in the myocardial tissues and ECM from patients with AF (Figure 1[E]). The WB results were consistent with those of the qPCR and IHC experiments (Figure 1[F]). In addition, as a downstream effector of SHP-1, we also examined STAT3 activation in the atrium. The phosphorylated form of STAT3 was clearly increased in the AF atrium relative to that in the SR atrium (Figure 1[F]). These results suggest that SHP-1 is downregulated, whereas STAT3 is upregulated, in samples from patients with AF.
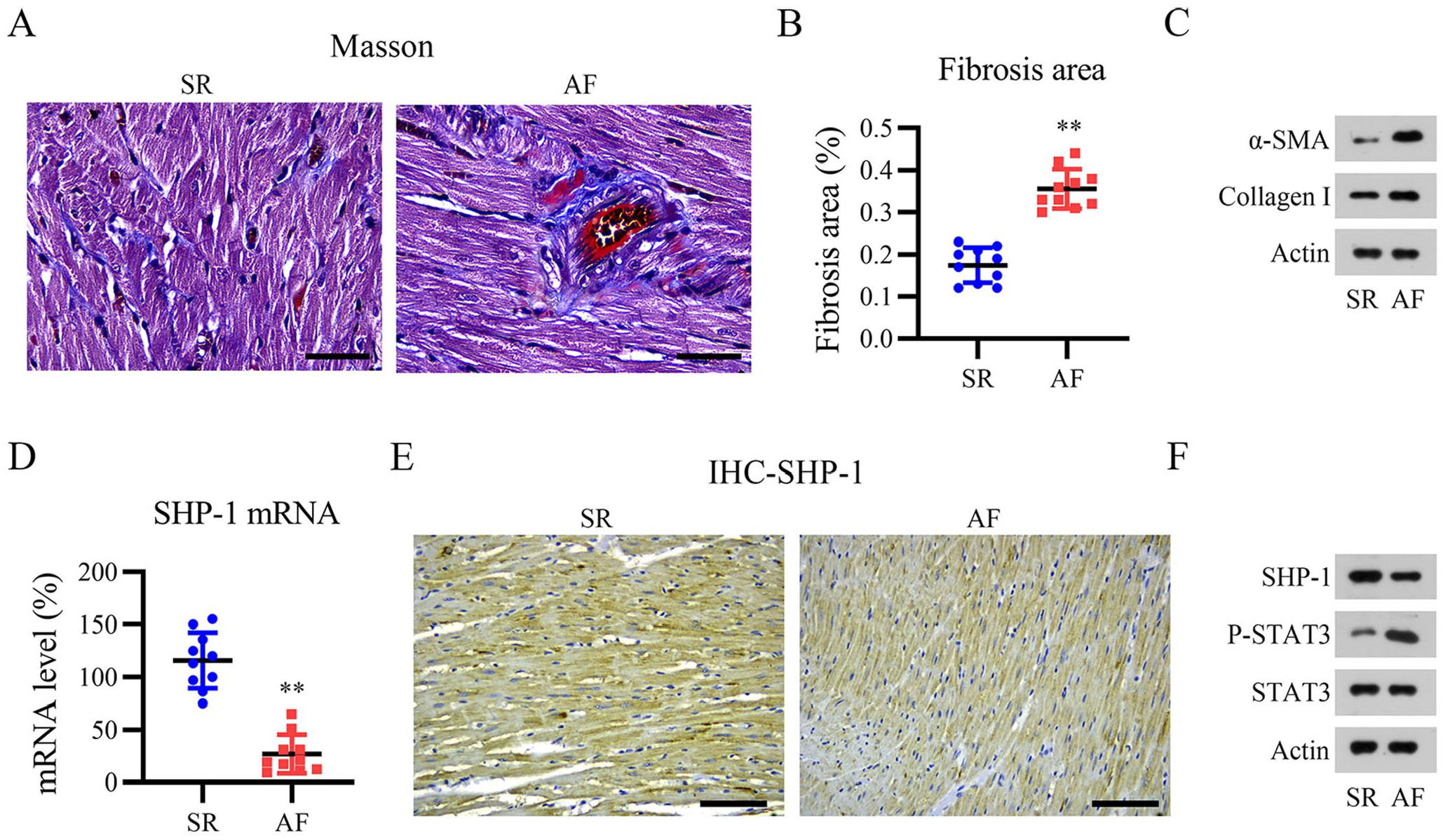
Decreased SHP-1 expression in patients with atrial fibrillation (AF) and severe atrial fibrosis. (A) Representative images of Masson’s trichrome staining. Scale bar, 200 μm. (B) Quantification of atrial fibrosis. (C) Western blotting (WB) analysis of α-SMA and collagen I levels in patients with normal sinus rhythm and atrial fibrillation. (D) SHP-1 mRNA levels were determined by quantitative PCR. (E and F) Immunohistochemistry (IHC) and WB analyses of SHP-1 protein expression. Scale bar, 200 μm.
SHP-1 is downregulated in AF mouse in vivo and in vitro models
To further evaluate the effect of SHP-1 on AF progression, we established in vitro and in vivo AF mouse models. First, mice were implanted with a minipump containing Ang II to induce AF. Compared with that in the sham group, SHP-1 expression was remarkably lower in AF mice, as assessed using qPCR, IHC, and WB (Figure 2[A] to [C]). Based on previous reports,19,20 we isolated both mouse myocytes and fibroblasts from the heart tissue of mice and treated these cells with Ang II to prepare an AF mouse cell model. The qPCR data showed that SHP-1 expression reached a minimum in myocytes treated with 4 μM Ang II and in fibroblasts treated with 8 μM Ang II (Figure 2[D] to [F]). WB showed that treatment with 8 μM Ang II downregulated SHP-1 expression in mouse myocytes and fibroblasts (Figure 2[E] to [G]). The WB results described above indicate that STAT3 phosphorylation was upregulated in the AF animal and cultured mouse cell models (Figure 2[C], [E] and [G]).
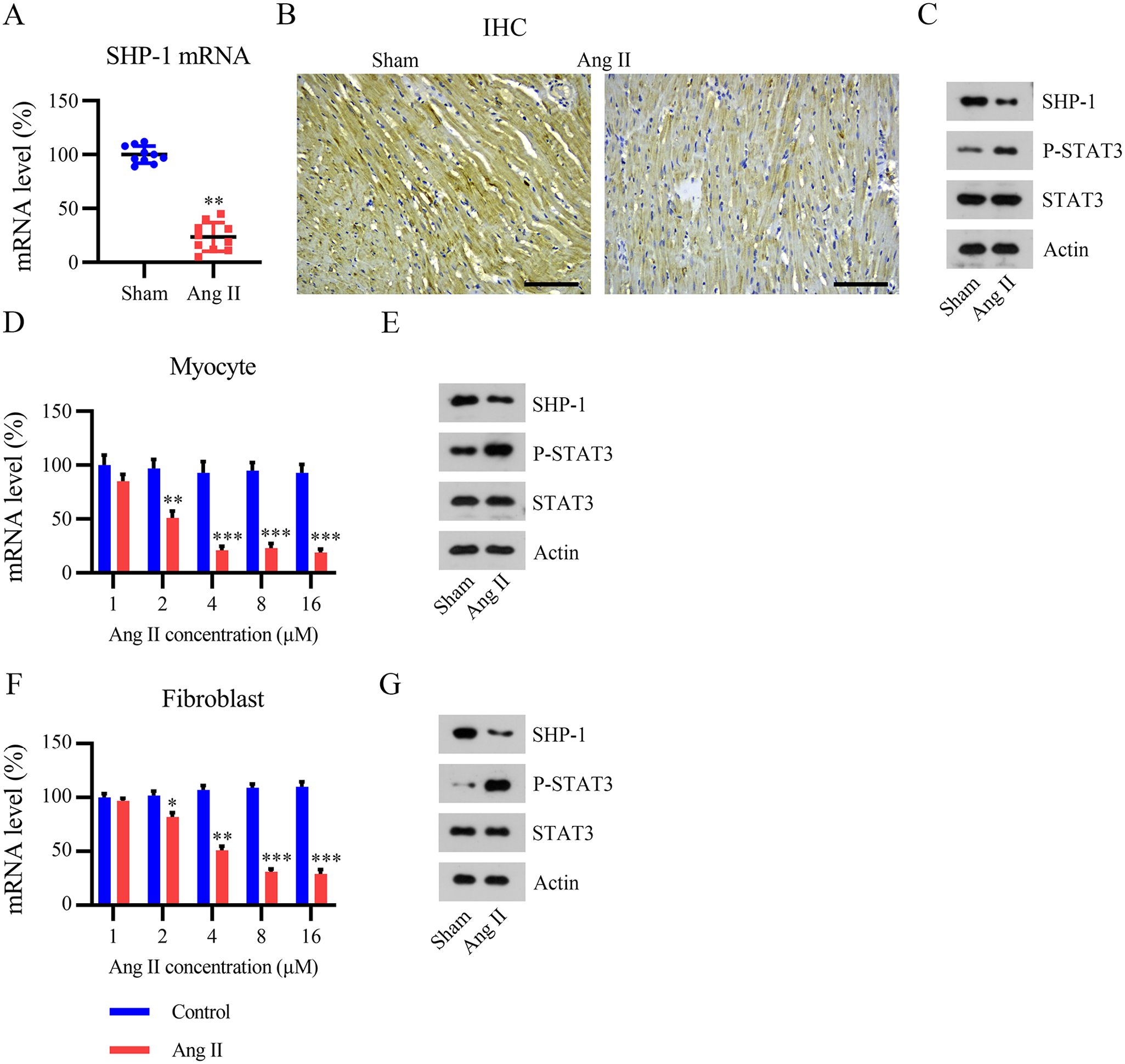
Expression of SHP-1 in angiotensin (Ang) II-induced mouse and cell models. Mice were implanted with a minipump containing Ang II to elicit AF. (A) SHP-1 mRNA level determined using qPCR. (B and C) IHC and WB analyses of SHP-1 protein expression. Isolated myocytes and fibroblasts were cultured with Ang II at the indicated concentrations for 12 h. Scale bar, 200 μm. (D and F) qPCR analyses of SHP-1 mRNA expression. (E and G) WB analyses of SHP-1 protein expression.
SHP-1 overexpression improved Ang II-elicited AF in vivo
To further evaluate the role of SHP-1 in AF, the LV-encoding SHP-1 gene was used to overexpress SHP-1 in mice with AF. Compared with the LV-NC infection group, LV-SHP-1 infection significantly increased SHP-1 mRNA and protein expression in Ang II-induced AF mice, as demonstrated using qPCR, IHC, and WB (Figure 3[A] to [C]). Furthermore, SHP-1 overexpression resulted in the dephosphorylation of STAT3 in the heart tissue of AF mice (Figure 3[C]).
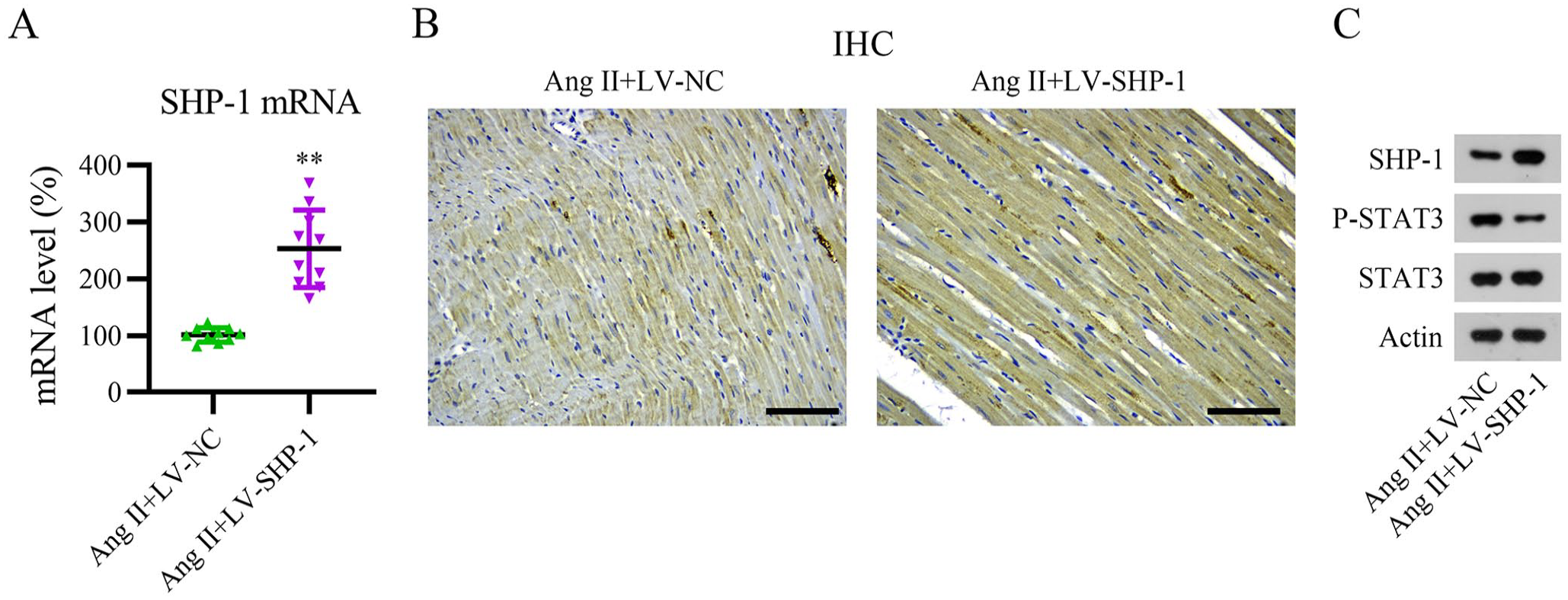
Overexpression of SHP-1 in heart tissue of an Ang II-induced mouse model. The pericardial space of mice was injected with lentivirus carrying negative control (LV-NC) or LV-SHP-1, and a minipump containing Ang II was implanted into mice to elicit AF. (A) SHP-1 mRNA levels were determined using qPCR. (B and C) IHC and WB analyses of SHP-1, STAT3, and phosphorylated STAT3 protein expression. Scale bar, 200 μm.
Subsequently, several AF indices and biomarkers were analyzed in Ang II-elicited AF mice, including the AF incidence and duration, IACT, and ERP. Figure 4(A) to (E) shows that Ang II administration markedly increased the AF incidence and duration as well as the IACT, reduced the ERP, and increased the expression of α-SMA and collagen I in mice, suggesting that the AF mouse model was successfully established. Moreover, Ang II infusion-elicited AF was remarkably improved in mice expressing LV-SHP-1 compared to that in mice expressing LV-NC. These results indicate that SHP-1 overexpression can improve Ang II-induced AF.
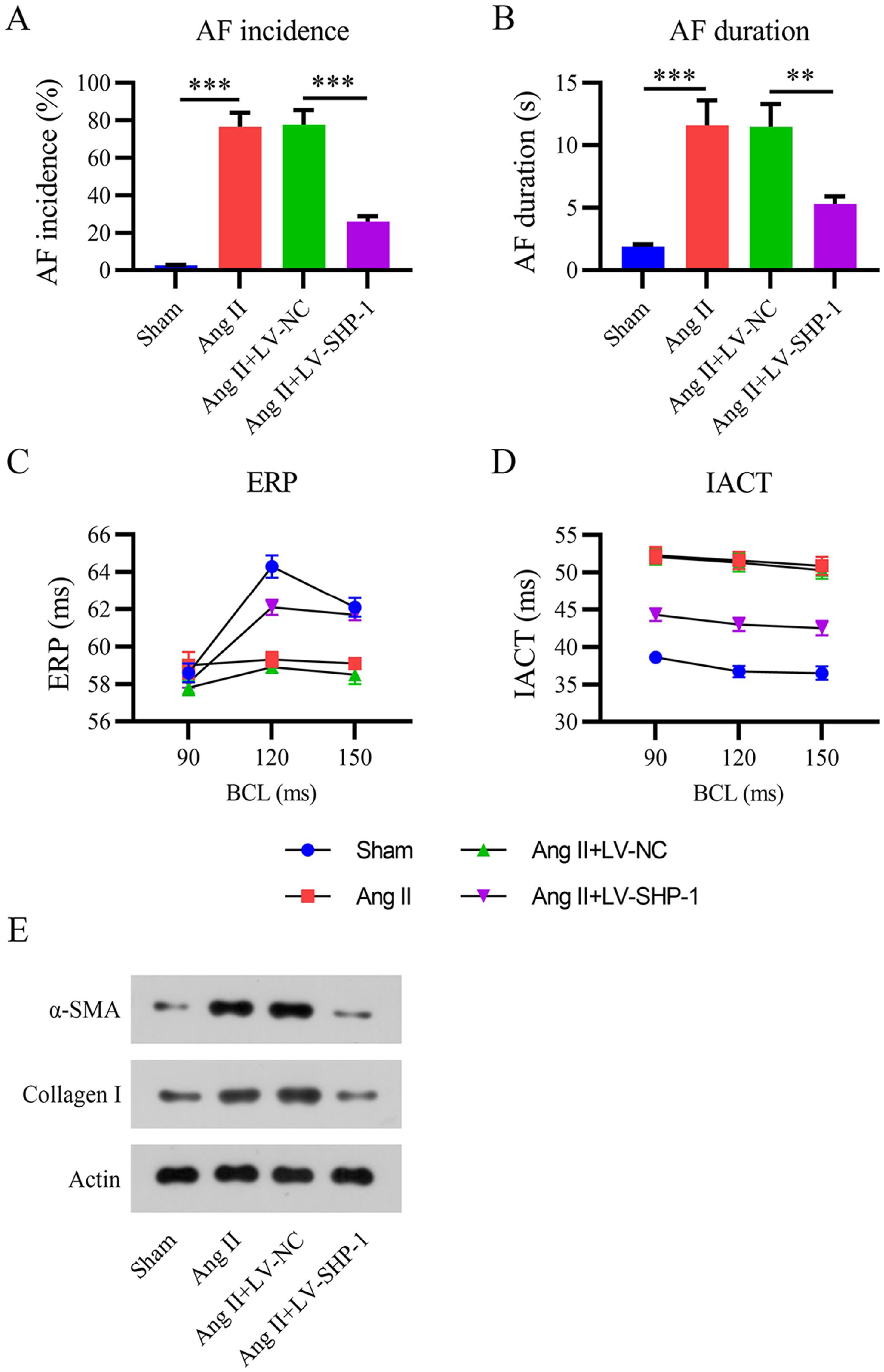
Overexpression of SHP-1 ameliorates Ang II-induced AF. (A) Effective refractory period (ERP). (B) Interatrial conduction time (IACT). (C) AF incidence. (D) AF duration at day 14. (E) WB analyses of α-SMA and collagen I protein expression. BCL: basic cycle length.
SHP-1 ameliorates Ang II-induced ROS elevation, fibrosis, and ECM deposition in an AF mouse cell model
To confirm the role of SHP-1 in our AF mouse cell model, we transfected Ang II-treated mouse myocytes and fibroblasts with an SHP-1 overexpression vector. Both qPCR and WB showed that transfection of the SHP-1 overexpression vector led to upregulated SHP-1 levels in Ang II-induced cells (Figure 5[A] to [D]). In addition, STAT3 phosphorylation was reduced in SHP-1-overexpressing cells (Figure 5[C] and [D]), suggesting that STAT3 was deactivated by SHP-1 in Ang II-treated cells.
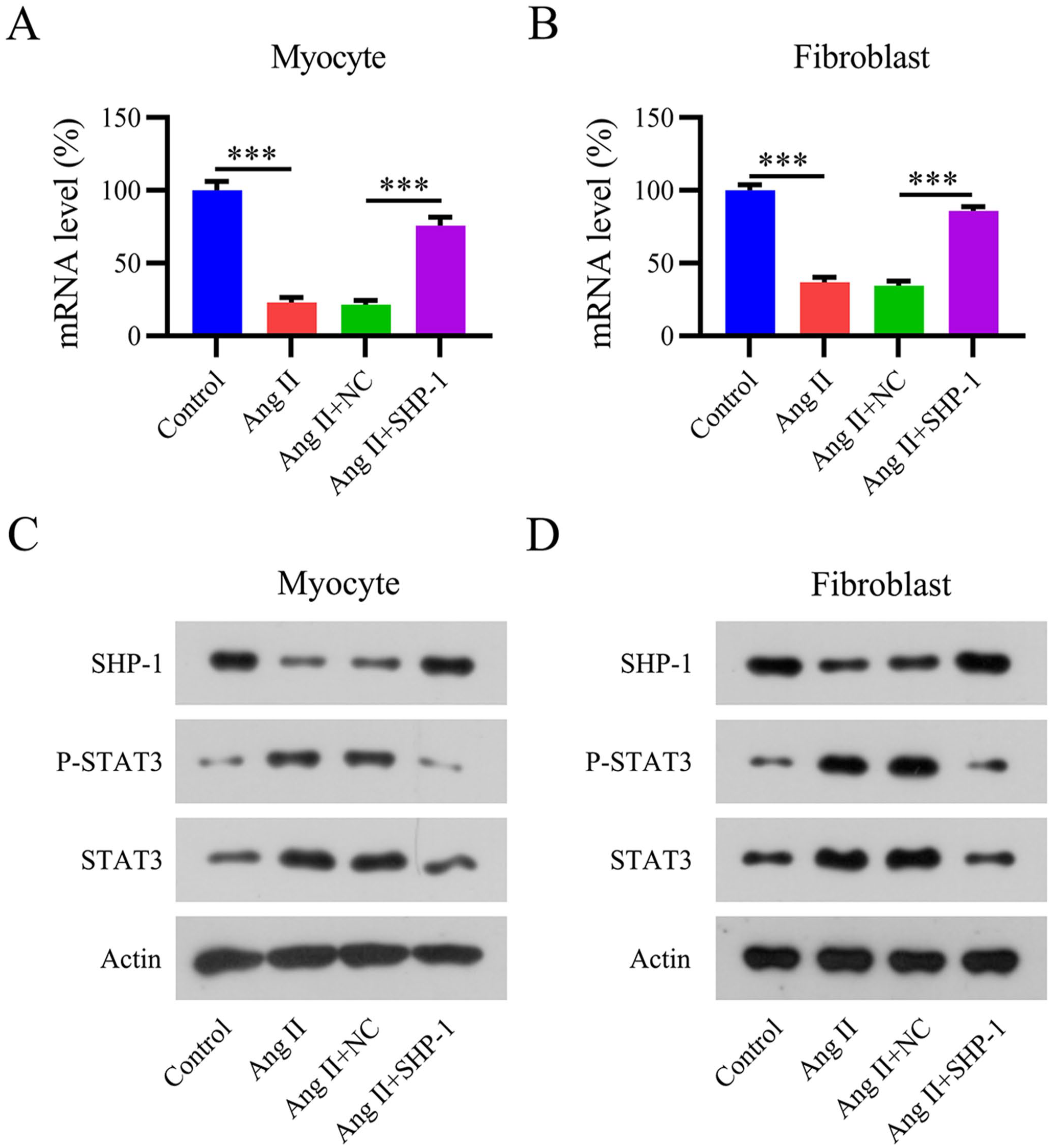
Overexpression of SHP-1 in an Ang II-induced mouse cell model. Isolated mouse myocytes and fibroblasts were transfected with an SHP-1 overexpression vector and cultured with Ang II at the indicated concentrations for 12 h. (A, B) SHP-1 mRNA levels determined using qPCR. (C, D) WB analyses of SHP-1, STAT3, and phosphorylated STAT3 protein expression.
Previous studies have indicated that ROS generation and ECM deposition are accompanied by AF progression and are the major mechanisms leading to myocardial fibrosis.21,22 Therefore, we examined ROS generation using flow cytometry and measured the expression of transforming growth factor beta 1 (TGF-β1)/mothers against decapentaplegic homolog 2 (SMAD2), collagen I/III, and matrix metalloproteinase 1 and 2 (MMP1/2) using WB. Ang II treatment induced excessive ROS production (Figure 6[A] and [B]) and activation of the fibrotic TGF-β1/SMAD2 pathway, as well as increased expression of collagen I/III and MMP1/2 in cells (Figure 6[C] and [D]). SHP-1 overexpression partially abolished these effects of Ang II, suggesting that SHP-1 ameliorates Ang II-induced ROS elevation, fibrosis, and ECM deposition in the AF mouse cell model.
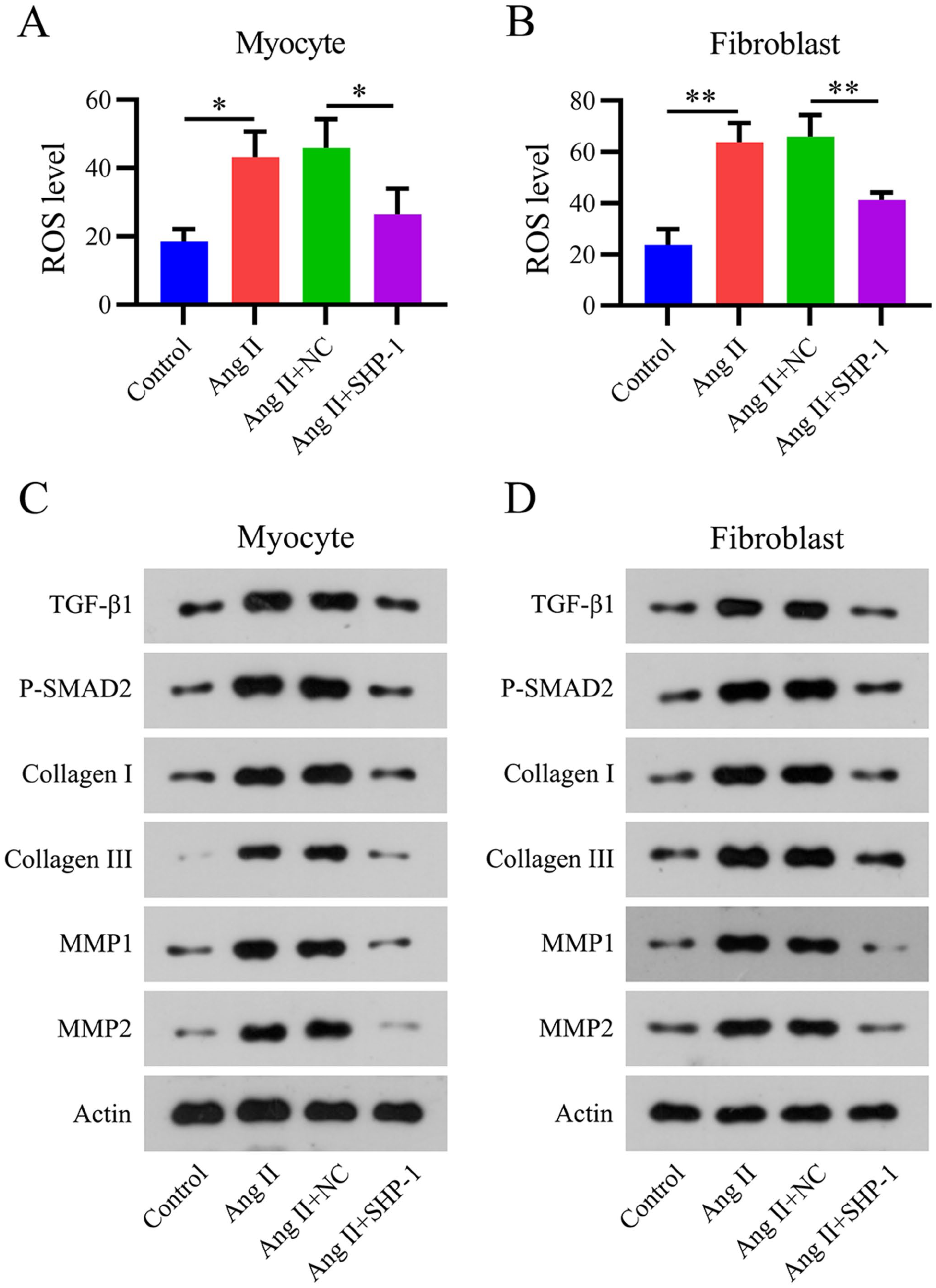
Overexpression of SHP-1 reduces reactive oxygen species (ROS) generation and extracellular matrix (ECM) deposition in an Ang II-induced mouse cell model. (A and B) Flow cytometry analysis of ROS levels in an Ang II-treated mouse cell model. (C and D) Expression of TGF-β1, phosphorylated SMAD2, collagen I/III, and MMP1/2 as detected by WB.
STAT3 reactivation counteracts the effect of SHP-1 overexpression on fibrosis development in an AF mouse cell model
To assess whether SHP-1 modulates STAT3 activation in an AF mouse cell model, Ang II-treated mouse myocytes and fibroblasts overexpressing SHP-1 were treated with 5 μM colivelin to reactivate STAT3. WB showed that colivelin administration elevated the protein levels of phosphorylated STAT3 in SHP-1-overexpressing AF mouse cells, whereas the expression of SHP-1 was not altered by colivelin (Figure 7[A] and [B]).
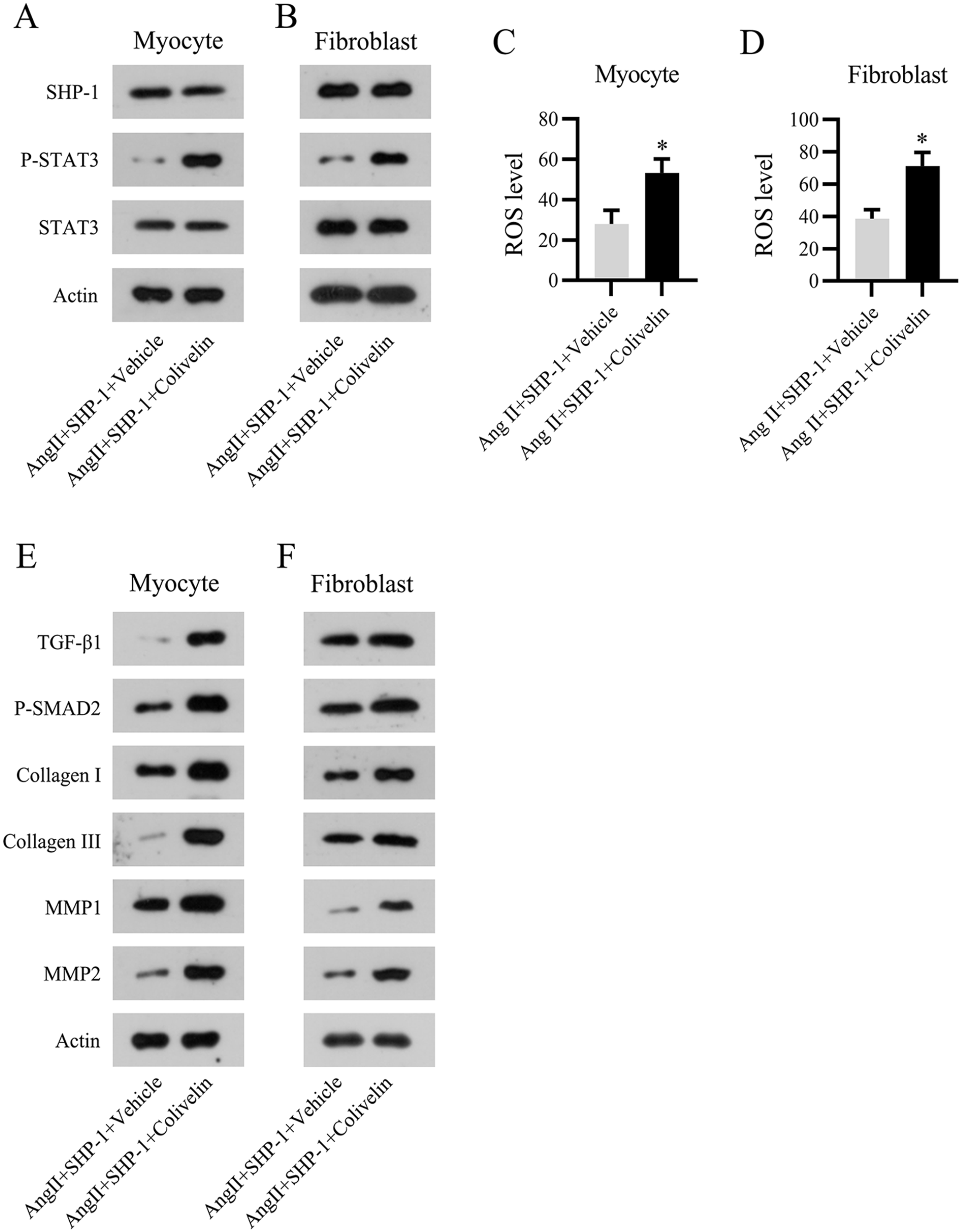
Reactivation of STAT3 abolishes the effect of SHP-1 in an Ang II-induced mouse cell model. Isolated mouse myocytes and fibroblasts were transfected with an SHP-1 overexpression vector and cultured with Ang II and colivelin for 12 h. (A) WB analyses of SHP-1 and STAT3 protein expression and phosphorylation of STAT3. (C and D) Flow cytometry analysis of ROS levels in the Ang II-treated cell model. (E and F) Expression of TGF-β1, phosphorylated SMAD2, collagen I/III, and MMP1/2 as detected using WB.
ROS generation was evaluated using flow cytometry. The results indicated that STAT3 reactivation led to excessive generation of ROS in SHP-1-overexpressing AF myocytes and fibroblasts (Figure 7[C] and [D]). Furthermore, activation of the TGF-β1/SMAD2 pathway and expression of the ECM proteins collagen I/III and MMP1/2 in AF mouse cells were promoted by colivelin (Figure 7[E] and [F]). These results suggest that SHP-1 overexpression affects fibrosis development in a STAT3-dependent manner in AF mouse cells.
Discussion
Despite evidence that high levels of SHP-1 may repress cardiac damage caused by various cardiovascular diseases, such as myocardial infarction, the mechanism of how SHP-1 modulates atrial fibrosis in patients with AF is unclear. In this study, patients with AF exhibited lower SHP-1 levels and dramatically higher levels of fibrotic factors compared to patients with normal SR. In addition, Ang II administration in mice and cell models led to low SHP-1 expression. Furthermore, SHP-1 overexpression ameliorated Ang II-induced AF in mice. In Ang II-treated mouse myocytes and fibroblasts, SHP-1 reduced ROS accumulation, deactivated the TGF-β1/SMAD2 pathway, and decreased the expression of collagen and matrix metalloproteinases (MMPs). We also found that activated STAT3 was negatively correlated with SHP-1 expression in AF mice and our cultured mouse AF cell model, and that STAT3 counteracted the SHP-1-induced inhibition of atrial fibrosis.
Atrial fibrosis is an obvious manifestation of the structural remodeling that occurs in patients with AF and forms the structural foundation for AF maintenance. Pathological alterations relevant to atrial fibrosis mostly result from aberrant collagen metabolism, an imbalance in collagen I/III levels, elevated collagen deposition, and fibrotic diseases.23,24 Ryu et al. 25 demonstrated that SHP-1 inhibits collagen release by inhibiting collagenases 1 and 3 and nitric oxide products modulated through nuclear factor kappa B (NF-κB)/p38 kinase. Here, we demonstrated that upregulation of collagen I/III and MMP1/2 induced by Ang II in both mice hearts and cell models was reduced by SHP-1 overexpression, confirming that SHP-1 inhibited atrial fibrosis.
TGF-β is regarded as a major mediator of atrial fibrosis.2,5 It activates atrial fibroblasts to differentiate into myofibroblasts and facilitates their proliferation, migration, and the production of ECM proteins. 26 In transgenic mice overexpressing cardiac TGF-β1, the atria showed obvious atrial fibrosis and AF properties despite equivalent TGF-β1 expression in the atria and ventricles.27,28 In our Ang II-induced mouse cell model, TGF-β1 expression was increased in both mouse myocytes and fibroblasts, whereas SHP-1 overexpression inhibited TGF-β1 expression, thereby repressing phosphorylated SMAD2 levels. These data suggest that SHP-1 inhibits the TGF-β1/SMAD2 pathway to suppress atrial fibrosis during AF progression.
Accumulating evidence strongly indicates that the Ang II-elicited STAT3 pathway is an essential component of AF pathogenesis.29 –32 In addition, in the non-Ang II-elicited AF model, STAT3 was associated with AF progression. For instance, fibroblast growth factor 23 (FGF23) can mediate AF via the STAT3 and SMAD3 pathways. 33 TGF-β has also been shown to contribute to AF through the STAT3 pathway. 34 MicroRNA-21 accelerates cardiac fibrosis progression via the STAT3 pathway; therefore, microRNA-21 suppression may improve cardiac fibrotic remodeling and AF because AF eventually develops into cardiac fibrosis. 35 Liu et al. previously studied the effect of SHP-1 in AF using the mouse atrial myocyte line HL-1. Overexpression of SHP-1 suppressed the activation of its direct binding partner c-Src, thus enhancing the transcriptional activation of Cx43, which strengthens gap junctions, thereby inhibiting AF. Our results are consistent with those of previous studies showing that SHP-1 inhibits AF progression 36 by directly influencing STAT3 downstream, which further deactivates the STAT3 pathway, MMPs, and accumulation of ECM components, such as collagen I and III.
In this study, we applied multiple complimentary methods to study the potential role of SHP-1 as a cardioprotective agent in preventing fibrosis and AF, using both human biopsies and mouse models to explore the underlying mechanistic changes associated with SHP-1, STAT3, fibrosis, and AF. However, there were some limitations to this study. First, we used LV infection-driven SHP-1 overexpression to elucidate the importance of the role of SHP-1 in AF mice; however, transgenic SHP-1-overexpressing mice would be preferable for these experiments. An SHP-1 knockdown/knockout mouse would also be a valuable tool for further studies. In addition, the role of STAT3 in SHP-1-mediated AF was not elucidated in vivo. Furthermore, the results of our mouse cell-based experiments should be validated in myocytes/fibrocytes of human origin.
In conclusion, SHP-1 is inversely related to AF severity, and its overexpression represses AF by dephosphorylating STAT3, leading to reduced atrial fibrosis, ROS generation, and ECM deposition. Therefore, overexpression of SHP-1 may improve AF, highlighting the importance of this molecule and its related pathways as promising targets for AF treatment.
Supplemental Material
sj-pdf-1-ebm-10.1177_15353702231165717 – Supplemental material for SHP-1 alleviates atrial fibrosis in atrial fibrillation by modulating STAT3 activation
Supplemental material, sj-pdf-1-ebm-10.1177_15353702231165717 for SHP-1 alleviates atrial fibrosis in atrial fibrillation by modulating STAT3 activation by Xiaobiao Zang, Zhihan Zhao, Ke Chen, Weifeng Song, Jifang Ma, Haixia Fu, Xianqing Wang and Yonghui Zhao in Experimental Biology and Medicine
Footnotes
Authors’ Contributions
All authors participated in the study design, analysis of the data, and review of the manuscript: XZ, ZZ, KC, WS, JM, HF, XW, and YZ conducted the experiments, and ZZ and YZ wrote the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: this work was supported by the Natural Science Foundation of China (grant no. U1504802), science and technology project of Henan Province (grant no. 222102310065), Joint Construction Project of Henan Medical Science and Technology (grant no. LHGJ20210091), and Joint Construction Project of Henan Medical Science and Technology Research Plan (grant no. LHGJ20200080).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.