
Abstract
Major depressive disorder (MDD) is a complex illness that is arising as a growing public health concern. Although several brain areas are related to this type of disorders, at the cellular level, the parvalbumin-positive cells of the hippocampus interplay a very relevant role. They control pyramidal cell bursts, neuronal networks, basic microcircuit functions, and other complex neuronal tasks involved in mood disorders. In resistant depressions, the efficacy of current antidepressant treatments drops dramatically, so the new rapid-acting antidepressants (RAADs) are being postulated as novel treatments. Ketamine at subanesthetic doses and its derivative metabolites have been proposed as RAADs due to their rapid and sustained action by blocking N-methyl-
Keywords
Impact statement
Depression is a common mental disorder that is becoming a public health problem worldwide. In this regard, the search for new treatments becomes highly relevant, and it has been recently described that blockade of N-methyl-
Introduction
Major depressive disorder (MDD) is a growing public health concern. The World Health Organization (WHO) ranked MDD as the third leading cause of illness worldwide in 2008, and it is conceivable that it will rank first by 2030. 1 MDD is a complex disease in which genetic susceptibility to lifestyle plays a crucial role. 2 MDD causes changes in mood, behavior, and memory and induces anxiety, nervousness, exhaustion, and stress among other symptoms that severely limit psychosocial functioning and reduces quality of life. 3 Therefore, predisposing factors to the illness produce disability and in some cases leads to suicide. 1 The diagnosis of neuropsychiatric disorders has increased markedly, by more than 22%, following the COVID-19 pandemic. 4 People who have overcome the disease have suffered from delusions, manic symptoms, poor memory, extreme fatigue, anxiety, and insomnia. 5 In practice, the diagnosis and management of MDD pose a challenge for clinicians due to its different presentations, unpredictable course, and prognosis, in addition to a highly variable treatment response. 3
Limbic system brain areas such as the medial prefrontal cortex (mPFC), amygdala (AMG), and hippocampus (HC) are among the brain areas affected in MDD. 6 Indeed, the HC plays a direct role in declarative memory being a key modulator of prefrontal function, cooperating in the regulation of explicit memory through its connections with the dorsolateral prefrontal cortex (dPFC). In addition to its involvement in learning and memory, the HC is also related to chronic pain management and emotional processes, mainly through close connections with the infralimbic area (IL) located in the mPFC. 7
Several studies have described a reduction in hippocampal volume in patients with chronic depression, 8 which in turn is related to neuronal and glial loss or cell atrophy. 9 Reduced levels of brain-derived neurotrophic factor (BDNF) in patients with MDD cause loss of dendritic spines of pyramidal cells (PCs) and thus decreased neuroplasticity. 10 These PCs are tightly interconnected with different subpopulations of interneurons (INs), establishing multiple connections with them. 11 The long-range inputs and outputs of PCs link the HC to several brain areas related to mood disorders, such as the frontal cortex and the anterior cingulate cortex.12,13 Thus, INs are known primarily because they establish and control the local neuronal population, but it is noteworthy that long-range inhibitory INs (parvalbumin [PV]- and somatostatin [SST]-positive) have also been described between the HC and AMG, and basal ganglia and neocortical areas. 11
The lack of the referred neuronal connectivity contributes to MDD.
14
In order to mitigate this disorder, many rapid-acting antidepressants (RAADs) are emerging in recent years as a promising therapy, including Ketamine, an N-methyl-
Hippocampal connectivity and MDD
The hippocampal formation, constituted by Cornu Ammonis (CA1, CA2, CA3, and CA4), Dentate Gyrus (DG), and Subiculum (SB), is profusely interconnected participating various types of neurons organized in different layers. The inputs come from cortical layer II/III via the perforant pathways (PPs) or temporo-ammonic pathway (TA), reaching the unmyelinated mossy fibers of the DGs located in the granular layer or the Schaffer collaterals in CA3, respectively. CA3 transmits information to CA1, which in turn sends projections to neurons of the entorhinal cortex (EC), located mainly in deep layers V and VI. 16 This complex neural circuitry is part of Papez’s network of memory or/and emotional circuits. 17 In addition, CA1 neurons project through the fimbria and fornix to return processed information to mammillary bodies located in the hypothalamus. They are also crucial of the neuroendocrine axis (hypothalamic-pituitary-adrenal, HPA) and play a key role in pathophysiology of mood disorders. 18
On the contrary, there are connections along the ipsilateral longitudinal fasciculus (ILF) with ipsilateral temporal and occipital areas. Likewise, several connections also extend to contralateral areas, including in greater numbers the long-range input PV INs. These long-projection GABAergic neurons reach the EC and medial septum (MS) via the retrosplenial cortex, exerting potent regulation of behavioral control over local hippocampal circuits through numerous disinhibitory synaptic mechanisms. 19
As mentioned above, impaired hippocampal function has been related to MDD. The efferent pathways of the subiculum and HC connect to critical areas such as nucleus accumbens (NAc) and ventral tegmental area (VTA) involved in the dopaminergic neurotransmitter-mediated reward loop, leading to alterations in the reward circuitry and affecting cognition and enjoyment. It has also been hypothesized that an excitatory dopaminergic projection loop between VTA and HC would enhance long-term potentiation (LTP), and in consequence, learning and memory. 20 Therefore, impairment of this hippocampal function could lead to reduced dopaminergic tone and contribute to anhedonia in MDD. 21 Finally, bed nucleus of the stria terminalis (BNST), located in the basal forebrain, has gained relevance because of its close relationship with the basolateral amygdala (BLA) which through the stria terminalis modulates the emotions of anger and fear. In addition, BNST, which presents sexual dimorphic structure, has recently been identified as an integrative center of limbic information. It presents connections with the AMG and also between the HC and the hypothalamic paraventricular nucleus (HPV). It has been observed that direct glutamatergic inputs from the ventral subiculum reached BNST, while outputs from the anterolateral, anteromedial, ventral, and posterior nuclei of the BNST also connect with the HC. Consequently, damage to the HC results in attenuation of the HPA loop and impaired hormone secretion. Therefore, this dysregulation contributed to the abnormal stress response observed in MDD. 22
Finally, a decrease in neurotransmitter release has also been described in MDD. 23 This alteration leads to synaptic dysfunction which in turn translates into morphological changes in neurons and has been described in brain areas such as the HC and the mPFC.24,25 In fact, the decrease of glutamate, serotonin, noradrenaline, or dopamine, among others, produces a lower activation of synaptic receptors accompanied by a drastic decrease of neurotrophic factors such as BDNF. The lack of monoamines and glutamate neurotransmitters release in the synaptic cleft could lead to down-regulation of excitatory synapses in multiple brain areas, 26 disrupting the HC and NAc and leading to several dysfunctions, mainly in reward circuits. 20 These patients suffer profound behavioral changes due to discouragement and fall into severe depressive states.
Role of hippocampal INs in MDD
INs represent 20–30% of the total neurons in the central nervous system (CNS) and act on the control of the excitatory firings, plasticity, synchronization of cortical rhythms, and maintenance and modulation of circuits. 11 Considering the very precise function performed by INs, a possible impairment of these neurons can lead to several pathologies such as schizophrenia, depression, or anxiety.27,28 In the HC, the correct activity of neural circuits is highly controlled by different types of GABAergic INs that constitute 7−11% of all hippocampal neurons. 12 PV, SST, and 5-HT3aR vasoactive or no vasoactive intestinal peptide (VIP/non-VIP) distinguish this population of hippocampal INs. Forty percent of the INs are PV-positive, whereas the SST and 5-HT3aR populations constitute 30% of hippocampal INs 11 (Figure 1).
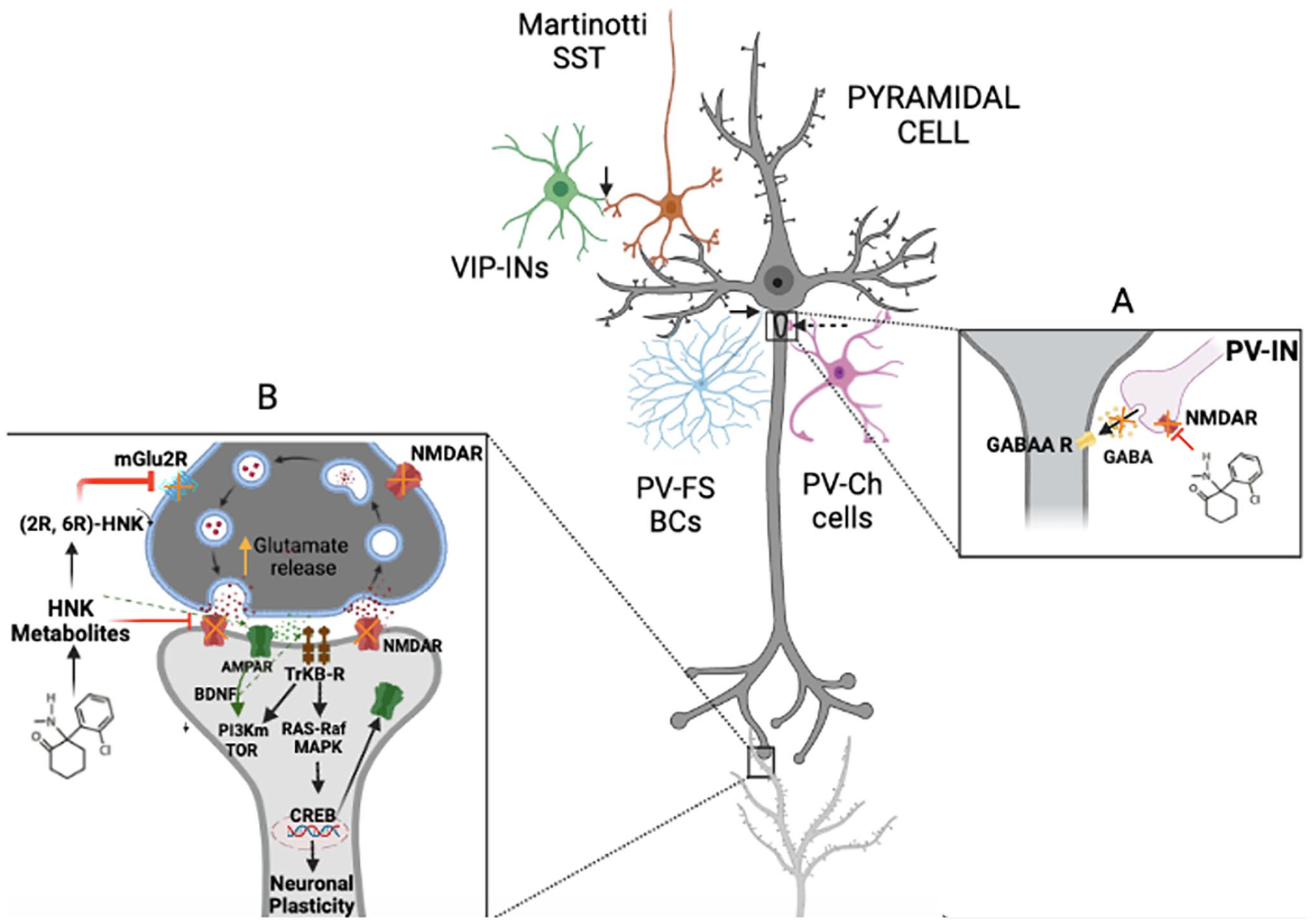
Representation of the action of ketamine on NMDARs and its effect on cellular synapses and activation of molecular pathways. Ketamine blocks NMDAR in high-affinity neurons, including PV-positive interneurons. Blockade of NMDARs prevents the release of the neurotransmitter GABA (A: right square) and consequently allows PCs to release more of the excitatory neurotransmitter glutamate (evoked release) (B: left square). These changes in the synaptic cleft induced an increased activation of AMPA receptors, which in turn promotes BDNF release. BDNF binds tightly to TrkB receptors promoting neuronal plasticity by activating the Akt-mTOR pathway cascade. The metabolite HNK appears as the last metabolite of ketamine, which has the ability to bind to AMPA receptors, potentiating BDNF and plasticity. Therefore, it represents a new step with potent antidepressant action. Continuous arrows show somatostatin Martinotti cells (SST) (orange) and VIP interneurons (green). SST targets dendrites, while VIP interneurons are selectively attached to SST. Non-VIP interneurons are not represented. Dashed arrows show parvalbumin interneurons (PV-INs) synapsis. Parvalbumin basket cells (PV-FS-BCs) (light blue) target PCs (dark gray) soma, whereas chandelier cells (PV-Ch cells) (purple color) target the axonal hillock.
Positive PV neurons are divided into two types: fast-spiking basket cells (FS-BCs) and chandelier cells (ChCs). Both types of cells synapse with PCs although they differ in their location. While BCs connect to the neuronal soma, ChCs synapse mainly directly with the axonal hillock. 29 Once inputs are received, action potentials are transmitted to the initial segment of the axon. 30 Although both types of PV+ INs are capable of producing a powerful influence on target neurons 31 (Figure 1(A)), FS-BCs show higher speed and temporal precision and are therefore specialized for fast excitation. 32 Also, FS-BCs are enriched with very high amount of sodium, Ca2+ permeable AMPA channels, 33 and excess of fast repolarizing Kv3 channels in fast-spiking axon neurons.11,34
Positive PV INs control basic microcircuits such as feedback and feedforward inhibition, playing a central role in the activity of complex neural network in the EC with high firing rates. 35 Moreover, in this context, FS-PV neurons are involved in plasticity 36 and stimulate reward behaviors in the mPFC. Therefore, they become the main target of emerging therapeutic treatments 37 appearing to be especially important in MDD.
In all anatomical structures of the HC, there are intimate connections between PCs and INs through performant pathways that exert tight functional control, especially in the DG. PV-expressing INs reach the granular layer of the DG and activate very important feedback and feedforward inhibition of the dentate mossy fibers located in the granular layer, 38 providing inhibitory control of the PCs (Figure 1(A)), evoking GABAergic postsynaptic currents and allowing fast-spike phases with precise control of spike timing. 39 However, disruption of this homeostatic balance in depressive disorders affects downstream molecular cascades and leads to synaptic dysfunction, neuronal damage, and decreased neuroplasticity. 40
Rapid action antidepressants: ketamine as a novel treatment in MDD
Adequate antidepressant treatment should reverse the molecular and even morphological changes observed in the HC, which is one of the main structures involved in MDD, linked to other areas such as the PFC, AMG, or cingulate gyrus, all interconnected through long tracts. 41
The therapeutic approach most commonly used worldwide to deal with MDD is based mainly on selective monoamine reuptake inhibitors selective serotonine reuptake inhibitors (SSRIs) and serotonine and norepinephrine reuptake inhibitors (SNRIs). These drugs act on the synaptic cleft, preventing the reuptake of synaptic monoamines and therefore increasing the amount of monoamines, which in the clinic translates into an effective antidepressant action. 42 One of the limitations of this treatment is that it takes several weeks to achieve therapeutic effect and restore or alleviate symptomatology. 43 Another type of effective antidepressant drugs is monoamine oxidase inhibitors (MAOIs), which prevent the degradation of monoamines. Tricyclic antidepressants, such as amitriptyline, also prevent monoamine reuptake. They are prescribed in MDD, anxiety, bipolar disorders, and other pathologies such as neuropathies and pain management, but the reported side effects have been greater than monoamine reuptake inhibitors (MRI). 26 Furthermore, the efficacy of these commonly used antidepressants is variable, and more than one-third of patients remain depressed. These patients could eventually slip into resistant depression (RD), leading to MDD and worsening of the disease. 44 Consequently, there is still a need to find new strategies that can contribute to obtain rapid or immediate improvement in those patients with severe symptoms. In this field, RAADs have emerged in the last years, and ketamine has been postulated as a novel and very promising treatment for MDD. 12
Ketamine hydrochloride was synthetized by Calvin Stevens in 1962 and has been used as an intravenous anesthetic since 1970, producing thalamo-cortical dissociative anesthesia and analgesia. 45 In addition to its use in surgery as an anesthetic, it is also used as an analgesic therapy to manage chronic pain. 46 Moreover, it has also been commonly used as a social drug worldwide. 47 As far as MDD is concerned, ketamine could improve hippocampal function by modulating INs and thus excitatory transmission, in addition to maintaining, promoting, and recovering dendritic spine loss. 44
Pharmacodynamic properties of ketamine and its metabolites control glutamatergic excitatory responses by antagonizing NMDARs,48,49 and it has been tested as a promising antidepressant therapy.50,51 Ketamine in subanesthetic doses is able to block NMDARs in INs. The increased release of glutamate allows more intense binding of the neurotransmitter to α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs), thus promoting quickly sodium and potassium permeability, and the activation of voltage-gated calcium channels (VGCCs). These channels trigger the release of BDNF, which, through binding to the receptor tyrosine kinase B (TrkB), produces the translocation of Akt to the plasmatic membrane and promotes the downstream molecular cascade through the rapamycin-mTORC1 pathway. It also regulates cellular transcription of CREB (cAMP response element binding protein) to promote neuronal survival and neuroplasticity. 6 The second BDNF-TrkB signaling pathway is also able to induce activation of the MAPK/Erk mTOR complex, inducing neurogenesis and dendritic growth. 52
Therefore, the affinity of ketamine to bind to NMDARs leads to appropriate neurotransmitter release at the synaptic cleft resulting in increased BDNF release and consequently increased plasticity and neuronal survival, among others, in the HC. In fact, subanesthetic ketamine administration promotes glutamate evoked release in the synaptic cleft in PCs (Figure 1(B)), triggering firing and neurotransmission and exerting a tight control and information transfer on different brain areas like PFC and HC. 53 Activation of these pathways is crucial in MDD as several studies have shown that postmortem tissues from suicide victims 54 and serum samples from MDD patients showed very low amounts of BDNF, whereas antidepressant treatment results in an increase of these values 55 accompanied by an improvement of symptomatology. 56
In addition to the above pathway, other signaling pathways are involved in this complex process. The rapid action of ketamine also activates the calmodulin-dependent kinase eEF2K which is directly dependent on the amount of calcium and calmodulin amount. 57 When NMDARs are disrupted by ketamine administration, eEF2K is suppressed leading to eEF2 disinhibition. This promotes rapid and sustained protein synthesis and BDNF release and has therefore been proposed as a very effective mechanism to palliate stress and neuronal plasticity. 58 In fact, some authors have proposed eEF2K as the main molecular substrate that potentiates the fast-acting depressant action of ketamine that responds very rapidly during stress events. 59
Furthermore, dissociation of ketamine produces extensive metabolites such as norketamine, dehydronorketamine, hydroxyketamine, and hydroxynorketamine (HNK). Recent studies have described the final metabolite of ketamine (2S, 6S; 2R, 6R)-HNK as the most present in plasma analysis in humans 60 and offer a promising finding due to its potentiating action of rapid antidepressant effect without side effects. 61 However, the mechanism of action differs from the previously mentioned NMDAR activation, and acts directly by binding specifically to the AMPAR. Consequently, it results in the direct release of BDNF into the synaptic cleft and downstream activation of TrkB. Also, it has been found that only (2R, 6R)-HNK metabolite is able to increase the levels of phosphorylated P70S6 kinase in threonine 389 position. This is potentiated by a central regulator of the mTOR pathway, inducing neuroplasticity. 62
Nevertheless, to achieve the therapeutic effects of ketamine and recover the hippocampal cytoarchitecture and cognitive deficits in human patients, the dose and exposure time need to be determined. 63 The first ketamine trial in depressed patients was performed with a single dose of ketamine 0.5 mg/kg administered intravenously over 40 min. Significant results were found in the depressed group with respect to the placebo group, indicating that ketamine-induced potentiation and control of glutamatergic neurotransmission in human RDs were able to strongly ameliorate depressive symptoms for several hours, maintaining these effects over several days. 48 Since then, different trials have been conducted with subanesthetic doses for MDD in rodents and humans.
Recently, favorable results have also been described for an adjusted intranasal dose of the (S)-ketamine enantiomer (28–84 mg) administered twice weekly for two weeks in combination with oral antidepressant treatment in patients with RD. 64 Similar results were obtained with a single infusion of the same dose with racemic ketamine (0.5 mg/kg) for 40 min in humans. 65 On the contrary, an intraperitoneal ketamine dose of 20 mg/kg was necessary to achieve antidepressant actions in mice. 66 In contrast, very low doses of ketamine (3–10 mg/kg) injected once in rodents also showed behavioral changes accompanied by recovery of spinal density compared to wild-type rodents. 67 It also has been reported that intraperitoneal infusion of 3, 10, or 30 mg/kg ketamine produced an immediate increase in glutamate, glutamine, and GABA in the mPFC of mice. 68 In relation to the observed GABAergic deficits and loss of glutamatergic homeostatic state described in MDD, the application of subanesthetic doses of ketamine (3 mg/kg) has also shown positive effects both in behavioral test and at structural and molecular levels. 69
Finally, increased synaptogenesis after ketamine treatment has also been described in rodents. 70 Likewise, in vivo two-photon images taken in the mPFC showed increased dendritic density with a single dose of 10 mg/kg of ketamine. 71 Other authors demonstrated that a single subanesthetic dose in rodents is able to exhibit increased dendritic density in brain areas such as the PFC through mTOR signaling. Two hours after a low dose of ketamine, induction of spine formation could already be observed, and after 24 h, maintenance of spine stability was observed through neurotransmitter-induced excitatory postsynaptic currents (EPSCs). This demonstrates that ketamine was able to increase nerve impulse frequency and amplitude. 72
Effects of ketamine on PV+ INs
Imbalance of PV+ neurons is related to pathologies such as anxiety, depression, and also schizophrenia. 73 These GABAergic INs play a crucial role in the control of local microcircuits as they perform direct inhibitory synapses on excitatory somatic neurons. 74 In fact, PV+ fast-spiking INs are known to have faster excitatory postsynaptic firing patterns than PCs, being better recruited by excitatory inputs. 75 Because of these characteristics, it is suggested that an NMDAR antagonist such as ketamine modifies FS-PV cell receptors more rapidly than excitatory PCs. 76 Indeed, inhibition of GABA release by PV-positive INs in CA1 or subiculum restores recurrent seizures. 27 Therefore, NMDAR dysfunction in FS-PV+ INs could be the main target in the treatment of patients with RD, and the non-competitive allosteric modulator ketamine appears as a key regulator of NMDAR in FS-PV+ to reduce severe depressive symptoms. 77
Ketamine shows high affinity for Ca2+-related influx channels, such as NMDARs, whereas its final metabolite HNK is a potent modulator of AMPAR. The relationship between both receptors is one of the links for the prevention of MDD by ketamine. In fact, ketamine plays a decisive role in PV+ INs due to the affinity shown by ketamine for NMDARs present in this type of INs 78 and the large amount of glutamate receptor 1 (GluR1) containing Ca2+-permeable AMPARs. 33 Calcium influx involves the activation of downstream molecular pathways involving BDNF, and as previously mentioned, they provide potent and stable control over PV+ INs, providing synaptic modulation. 79
PV FS-BCs seem to respond more rapidly to ketamine because they present the highest number of NMDA2A subtype receptors, as well as much more than pyramidal neurons. 80 NMDA2A receptors appear predominantly in the synaptic cleft, whereas extra-synaptic receptors present other types of subunits such as NMDA2B/2C/2D. NMDA2B receptors are bound to Ca2+-dependent calmodulin kinase II (CaMKII), 81 and together with NMDA2A receptors are considered the most important receptor subtype for transmitting burst, which in turn accounts for the majority of Ca2+ permeability subtype receptors among NMDARs. 82 This is an essential point to increase the efficiency and speed of inhibitory output. 37 In fact, recent work has shown that a low dose of ketamine (8 mg/kg, i.p.) enhanced NMDA2A receptor-mediated cortical activity in PV+ cells. 83 Furthermore, electrophysiological studies demonstrated antidepressant actions through the disinhibition hypothesis. 84 Initially, low doses of ketamine (10 mg/kg, i.p) were administered 24 h prior to animal testing, which led to a rapid blockade of NMDA2B in GABAergic INs. Consequently, sustained excitatory postsynaptic currents were generated and recorded in layer V PCs together with the BLA and provided therapeutic effects to mice. Although different types of INs were involved in these neuronal circuits, all of them showed a high density of NMDA2B subtype receptors. In fact, a recent study has shown that ketamine has no antidepressant effects in the absence of this receptor on GABAergic INs in knockdown mice. 85
On the contrary, NMDA2D subunits are expressed in some PV INs in the neocortex, neostriatum, and HC. 86 Some authors have described that Mg2+ does not provide a special blockade of NMDA2D channels, giving electrophysiological properties of fast spiking compared to another class of INs.87,88 This evidence supports that ketamine and other NMDAR antagonists such as MK-801 act with especially high affinity on NMDA2DRs in FS-PV cells, also supporting the role of this INs in the hypothesis of tightly controlled disinhibition of PCs.
Positive PV neurons are also related to the correct generation of gamma waves in the HC. 89 These oscillations are critical for neural network maintenance and cognitive phenomena. 90 Altered gamma waves with involvement of PV+ cells have been described in mood disorders or schizophrenia. 91 In this regard, other authors demonstrated that a single dose of 10 mg/kg, i.p. ketamine administered for seven days was able to potentiate evoked action potentials in PV+ cells, counteracting the loss of axonal boutons induced in chronically stressed mice. 92 In this study, substantial evoked action potentials were also observed in PV+ INs 10 min after ketamine administration (10 μM, i. p). This effect was maintained for 50 min, supporting that ketamine binds NMDAR in PV+ INs increasing their excitability after stress exposure. 92
Conclusions
In conclusion, very low doses of ketamine, called hormesis, have shown a promising approach in MDD. The main activation pathway studied has been BDNF-TrkB-mTOR, promoting rapid neuronal plasticity. The changes in neurotrophic factors involve INs, and among them, PV+ INs play a pivotal role. They provide synaptic changes in different brain areas through the hypothesis of disinhibition, producing a rapid recovery in patients suffering from MDD. In only a few minutes, sustained antidepressant therapeutic effects are achieved during several hours or days.
Nevertheless, the beneficial effects of ketamine in MDD depend on the dose and time of exposure. While ketamine administered in a single dose may be very effective due to its action as RAAD, if administered repeatedly, it may produce stereotyped behaviors because of the loss of INs, and in particular, of PV+ neurons. Therefore, it is necessary to determine these variables for a safe approach to this promising treatment for MDD patients.
Footnotes
Acknowledgements
This manuscript is dedicated to the memory of Prof Felix-Cruz Sánchez on the twelveth anniversary of his passing.
Authors’ Contributions
JVL and PAG designed the project. IBA with the collaboration of MAG, AV-R, and MP-G researched data for the article and bibliography. Figure was designed by IBA in collaboration with NO and HB. IBA wrote the first draft. NO, HB, PAG, and JVL contributed to discussion of the content and reviewed and edited this paper before submission. All authors have read and agreed to the published version of the manuscript due to the knowledge and experience in the study of depressive disorders.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work has been partially supported by the Basque Government (GIC 21/133, IT 1706-22 and PIBA_2020_1_0048).