
Abstract
MicroRNAs (miRNAs) have been identified as crucial modulators of gene expression and to play a role in palatogenesis. The aim of this study was to explore the potential role and regulatory mechanisms of miRNAs during palatogenesis. RNA-sequencing was performed to compare the RNA expression profiles of mouse embryonic palatal shelf (MEPS) tissue between an all-trans retinoic acid (ATRA)-induced group and control group, followed by reverse transcription–quantitative polymerase chain reaction for validation, demonstrating upregulated expression of miRNA-470-5p and downregulated expression of Fgfr1 in the ATRA-induced group. The specific binding sites of miRNA-470-5p that potentially govern Fgfr1 expression were predicted by miRanda and TargetScan. The relationship between miRNA-470-5p and Fgfr1 was validated in HEK293T cells by luciferase reporter assays, confirming that miR-470-5p acts directly on the Fgfr1 3′-untranslated region. Fgfr1 mRNA and FGFR1 protein levels were markedly downregulated in MEPS epithelial cells over-expressing miRNA-470-5p. Functional experiments in vitro with CCK-8, cell colony formation, and 5-ethynyl-2′-deoxyuridine (EdU) staining assays revealed that upregulated miRNA-470-5p expression could inhibit the epithelial-mesenchymal transition (EMT) of MEPS epithelial cells by targeting Fgfr1. These findings provide a new molecular mechanism of cleft palate formation, which can inform the development of new treatment and/or prevention targets.
Impact Statement
MiRNA is considered as a potential modifier gene of human cleft palate, but the specific molecular regulation mechanism remains unknown. In this context, we showed that the miRNA-470-5p suppressed the epithelial-mesenchymal transition of embryonic palatal shelf epithelial cells by targeting Fgfr1 3′-untranslated region during palatogenesis in mouse model. The findings indicated that these molecular biomarkers could be applied to improve early screening and/or targets for prevention and treatment of cleft palate.
Introduction
Cleft palate (CP) is a prevalent congenital orofacial deformity in newborns, occurring in approximately 1 in 700 live human births with a gradually increased incidence worldwide each year.1,2 The etiology of CP remains poorly understood, involving various genetic and environmental factors and their interactions.3–5 Patients have a variety of clinical presentations, resulting in varying levels of disability and functional impairment, including speech and eating problems. Patients generally require multidisciplinary treatment from shortly after birth to adulthood, such as CP repair surgery, orthodontic treatment, and speech therapy.6,7 Despite the low mortality rate of infantile CP, lifelong physical and psychological problems may occur, which greatly reduce quality of life. 8 However, the detailed etiology and pathogenesis of CP have not yet been elucidated.
To date, numerous microRNAs (miRNAs) have been demonstrated to play key roles in various biological processes, including cellular differentiation, proliferation, and migration, by regulating target gene expression through binding to the reverse complementary paired sequences of the 3′-untranslated region (UTR).9–12 MiRNAs function as post-transcriptional repressors and are considered to be key regulating factors of embryonic development and pathogenic factors of disease. MiRNAs modulate gene expression through inducing translational suppression and destabilization of mRNAs at the post-transcriptional level. Increasing evidence demonstrates that the gene repression effects of miRNAs 13 play significant roles in development of the palatal shelves (PSs), eliciting strong research interest. For example, miR-200b, which is expressed in medial edge epithelial (MEE) cells during palate development, was reported to regulate the expression of Zeb1 and Zeb2, key genes for palatal epithelial fusion. 14 In addition, miR-17-92 was found to suppress the transforming growth factor-beta (TGFβ)-induced proliferation inhibition and collagen synthesis of palatal mesenchymal cells in mice, directly targeting several cell proliferation regulators, including Tgfrb2, Smad2, and Smad4, to ultimately control palatal development. 15 MiRNA-140 was shown to negatively regulate Pdgfra protein production in zebrafish during palate morphogenesis. 16 Notably, miRNAs are considered potential modifiers of human CP-associated genes and could thus serve as a novel means to understand the etiological mechanisms of CP. In this study, next-generation RNA-sequencing (RNA-seq) was employed to analyze the RNA profiles related to CP formation. Through differential expression analysis, we have identified miRNA-470-5p as a novel miRNA that acts directly on the target sequence of the Fgfr1 3′UTR. Fibroblast growth factor signaling has been shown to be essential for governing PS growth, and Fgfr1 mutation is one of the important causative factors in the etiology of CP. Currently, there have been few studies on miRNA-470-5p, especially in CP formation, which have not been reported.
One of the most important events leading to CP is the failure of embryonic palatal fusion. During palatogenesis, the PS on both sides of the intraoral space are raised to form the transient medial epithelia seam (MES). Adhesion of the PS and complete fusion of the MES is considered to be the critical stage during palate development. 17 Nawshad 18 showed that the epithelial-mesenchymal transition (EMT) is a pivotal mechanism of MES disintegration. Palatal fusion is caused by EMT of the MEE seam, 19 and failure in the EMT phase of palatogenesis is one contributor to CP formation. Palate development is tightly regulated through a precise gene regulation network that controls cell migration, growth, differentiation, and apoptosis.
All-trans retinoic acid (ATRA), a derivative of vitamin A, is a strong teratogen that can cause CP formation by disrupting any stage of palatogenesis, such as ATRA exposure on embryonic day 10 (E10). 20 In the present study, C57BL/6J mice were used to establish an in vivo embryonic CP model by ATRA induction. The expression profiles of miRNAs and mRNAs between ATRA-induced and control mice were obtained by small RNA-seq and mRNA-seq. Based on the RNA-seq results, the specific binding sites of differentially expressed miRNAs and candidate target genes were predicted by miRanda and TargetScan tools. However, target prediction remains highly challenging and may produce false-positive results. It is important to experimentally validate the potential interactions of predicted miRNA-mRNA pairs. In our study, we described a simple and reliable assay, dual-luciferase reporter assay, to detect specific miRNA effects on possible target genes. The expression levels were then validated using reverse transcription–quantitative polymerase chain reaction (RT-qPCR), and the interaction between the CP-associated miRNA and mRNA target was confirmed using dual-luciferase reporter assays. Western blotting was further used to verify the changes at the protein level. Moreover, functional in vitro assays were performed to evaluate the function of the candidate CP-associated miRNA and its relation to EMT in palatogenesis. These findings can provide new insight into the molecular pathogenic mechanisms contributing to CP formation.
Materials and methods
Animals and specimen collection
C57BL/6J mice (8–9 weeks old, ~20 g, n = 45 [15 males, 30 females]; Beijing Vital River Laboratory Animal Technology Co., Ltd. Beijing, China) were used to establish the in vivo embryonic mouse CP model by ATRA induction. At 9- to 10-weeks-old, two female mice were coupled with one male mice overnight. The female mice were then examined for vaginal plugs the following morning at 08:00. The presence of vaginal plugs was considered E0.5. Pregnant mice at E10.5 were treated with 70 mg/kg ATRA (Sigma-Aldrich Co., Jefferson, MO, USA), dissolved in corn oil by gavage. The control group was provided 0.20 mL corn oil without ATRA by gavage. At E13.5, pregnant mice were euthanized using CO2, and embryonic PS tissue was harvested from the embryos. The tissue was dissected out under a microscope and stored at −80°C for further study. This study was approved by the Animal Ethics Committee of Shantou University Medical College (SUMC2019-297).
Morphological observation of the palate shelf
The heads of ATRA-induced and control group embryos were harvested at E13.5. All samples were formalin-fixed and paraffin-embedded. The paraffin-embedded embryonic head was cut into 4-μm-thick continuous sections using a Leica HistoCore BIOCUT microtome (Shanghai, China). The sections were then stained with hematoxylin and eosin (H&E) according to standard procedures, 21 and the morphological changes were observed under an OLYMPUS BX51 industrial microscope (Tokyo, Japan).
Sequencing and data analysis
Total RNA was extracted from the PS of E13.5 C57BL/6J mouse embryos in the ATRA-induced and control groups using a mirVana™ miRNA Isolation Kit (Ambion-1561, Austin, TX, USA) according to the manufacturer’s instructions. The library was constructed using TruSeq Stranded Total RNA with Ribo-Zero Gold, and then sequenced using the Illumina HiSeq 2500 platform (San Diego, CA, USA). High-quality reads were filtered from the basic reads to ensure clean reads suitable for subsequent analyses. All clean reads were compared with sequences in the Rfam database (version 10.0) and then annotated to discern known mRNA and miRNAs. The DESeq R package 22 was used to normalize the counts, and differentially expressed mRNA and miRNAs were compared, calculated, and filtered according to the threshold of P < 0.05 and log2FoldChange (FC) > 1. The miRanda and TargetScan tools were used for target gene prediction. 23
RT-qPCR
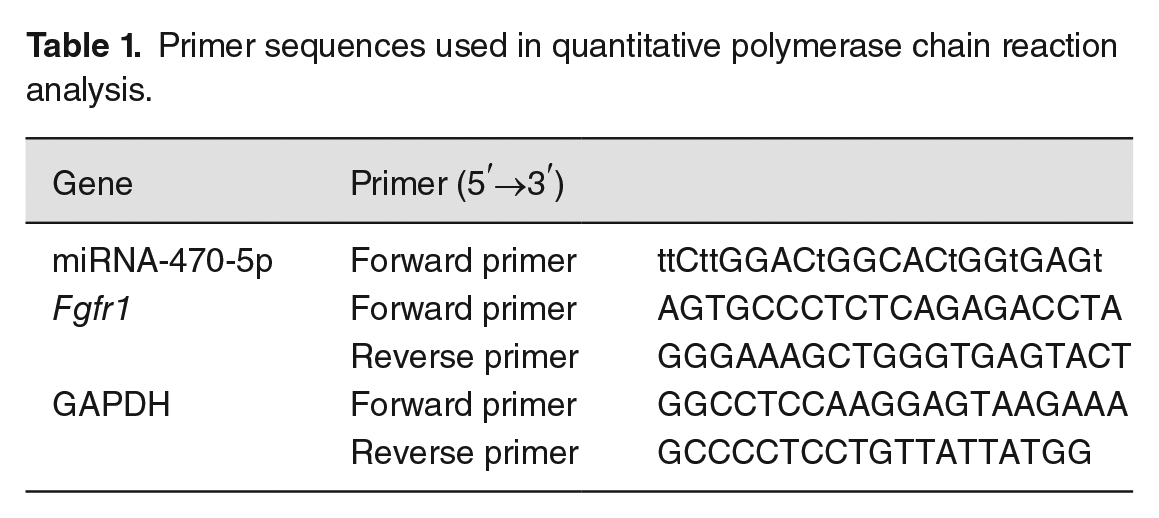
Total RNA from the PS of E13.5 C57BL/6J mouse embryos in the ATRA-induced and control groups was isolated using Trizol (15596-026; Invitrogen, Carlsbad, CA, USA) solution and then reverse-transcribed into cDNA using PrimeScript RT reagent kit (Takara Bio, Inc., Tokyo, Japan) according to the manufacturer’s specifications. Quantitative PCR was then performed using a DNA Engine Opticon II Continuous Fluorescence Detection System (CFX96, Bio-Rad, Hercules, CA, USA). The cycling conditions were as follows: cycle 1, 94°C (3–4 min); cycle 2, 94°C (30 s), 60°C (30 s), 72°C (30 s), repeated 30 times; cycle 3, 72°C (10 min). Gene expression levels were normalized to that of GAPDH using the 2-ΔΔCt method. 24 The GAPDH, miRNA-470-5p, and Fgfr1-specific primers (Qiagen, Venlo, Netherlands) are listed in Table 1.
Primer sequences used in quantitative polymerase chain reaction analysis.
Cell culture
Mouse embryonic palatal shelf (MEPS) epithelial cells were obtained from the medial PS of E13.5 mouse embryos. MEPS epithelial cells frozen in liquid nitrogen were thawed in a 37°C water bath. Then, the cell suspension was transferred to a 15 mL centrifuge tube and prewarmed culture medium was added dropwise to a total volume of 10 mL. The mixture was centrifuged, and the supernatant was removed. The remaining cell pellet was resuspended and incubated in high-glucose Dulbecco’s modified Eagle medium (DMEM) (Hyclone, Cat. No. SH30022.01B) supplemented with 10% fetal bovine serum (FBS; Hyclone, Cat. No. SH30087.01) and 1% penicillin streptomycin (Hyclone, Cat. No. SH30010), at 37°C in 5% CO2 and saturated humidity. Cells from passage 3 were used for subsequent experiments.
Luciferase reporter assay
According to the prediction of target genes through the miRanda (Figure 3(a)) and TargetScan (Figure 3(b)) databases, the wild-type (Wt) (5′-CCCAGGTGTTGGACCAAGACCCGCCCCGCT-3′) (mutation site, CCAAGA) or mutated (Mut) (5′-CCCAGGTGTTGGATTCCACCCCGCCCCGCT-3′) fragment of the Fgfr1 3′-UTR (Sangon Bioengineering Co., Ltd., Shanghai, China) was cloned into the psiCHECK™-2 vector (Promega, C8011). Human embryonic kidney 293T cells (HEK293T cells, Procell, Wuhan, China) were transfected with miRNA mimic negative control (miR-NC), miRNA-470-5p mimics, Fgfr1-Mut vector, or Fgfr1-Wt vector using Lipofectamine 2000 (Invitrogen, USA, 116680119), following the manufacturer’s protocol. The transfected cells were cultured in high-glucose DMEM with 10% FBS as described above, and the luciferase activity was measured after transfection for 48 h using a dual-luciferase reporter assay detection kit (Promega, E5331).
Cell transfection
miRNA-470-5p-inhibitor, miRNA-470-5p-mimic, and the corresponding negative control oligonucleotides were obtained from Guangzhou Ruibo Biotechnology Co., Ltd. (Guangzhou, China). According to the manufacturer’s specification, the above-mentioned plasmids and their corresponding negative controls were transfected into MEPS epithelial cells using Lipofectamine RNAiMAX (Invitrogen, Cat. No. 13778075). RT-qPCR was performed as described above 24–36 h after transfection, and Western blotting (as described below) was performed 36–48 h after transfection to measure the Fgfr1 mRNA and FGFR1 protein expression level, respectively.
Western blot analysis
At 36–48 h after transfection, proteins were extracted from MEPS epithelial cells with radioimmunoprecipitation assay lysis buffer (Merck KGaA, Darmstadt, Germany). The protein concentration was measured. Denatured samples were sequentially fractionated on a 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel and electro-transferred to a polyvinylidene difluoride membrane. Subsequently, the membrane was blocked with 5% fat-free milk and incubated with anti-FGFR1 antibody (A0082, ABclonal, Woburn, MA, USA) overnight at 4°C, followed by incubation with horseradish peroxidase-labeled secondary antibodies for 2 h at room temperature. The bands were visualized using BeyoECL Plus reagent (Beyotime, Beijing, China) and the intensities of the bands were quantified with Quantity One software (Bio-Rad, Hercules, CA, USA).
CCK-8 assay
The transfected MEPS epithelial cells were placed on 96-well plates. A Cell Counting Kit-8 (CCK-8) (Biyuntian, Shanghai, China) was used to measure cell proliferation at 0, 24, 48, and 72 h after transfection in accordance with the manufacturer’s instructions. The optical density values were measured at a wavelength of 450 nm using a microplate reader (Thermo Fisher Scientific, NY, USA).
Colony formation assay
The transfected MEPS epithelial cells were incubated in 96-well plates (NEST, Cat. No. PPP-001-030) at a density of 3000 cells/mL for 7 days. Any colonies that formed were then fixed in 4% paraformaldehyde and stained with 1% crystal violet for 20 min. The total number of colonies was counted, and plates were imaged using an enzyme-linked immunospot reader (AID Ispot system, Strassberg, Germany).
EdU staining
EdU (5-ethynyl-2′-deoxyuridine) staining was carried out using an EdU kit (Ruibo, Biotech, Gunagzhou, China, C10314) according to the manufacturer instructions. The EdU solution was seeded into the epithelial culture medium for incubation at 37°C for 2 h. The transfected MEPS epithelial cells were added to cell fixation solution (phosphate-buffered saline [PBS; Gibco] containing 4% paraformaldehyde) and incubated for 15–30 min at room temperature. The MEPS epithelial cells were then incubated with glycine for 10 min at room temperature and permeabilized with PBS containing 0.5% TritonX-100 for 5 min. The cells were incubated with Apollo reaction mixture, shielded from light, and left to react for 30 min at room temperature. Finally, the cells were incubated with Hoechst 33342 solution in the dark for 10 min to stain the nuclei. The images were captured using a Leica DMI6000B microscope (Wetzlar, Germany). Proliferation was measured by calculating the ratio of EdU-positive to DAPI (4′,6-diamidino-2-phenylindole)-stained cells.
Statistical analysis
SPSS (V26; IBM, Armonk, NY, USA) and GraphPad Prism (V7; La Jolla, CA, USA) were used for data analyses. Statistical comparisons of two groups were performed by independent t-tests. Unsupervised hierarchical clustering analysis was implemented to calculate the differential mRNA/miRNA expression levels. Results were considered statistically significant at P < 0.05 and absolute log2FC value > 1.
Results
Morphological and histological characteristics of the MEPS
PS tissues were obtained from the embryos of C57BL/6J mice at embryonic day 13.5 (E13.5) and stained with H&E for morphological observation. The width of the bilateral PS seam in the ATRA-induced group was significantly larger than that in the control group (Figure 1(a) and (d)). In addition, the elevation of the bilateral PS above the dorsum of the tongue and MEE fusion in the anterior and posterior domains were slightly delayed in the ATRA-induced group (Figure 1(b) and (c)) compared with those of the control group (Figure 1(e) and (f)).
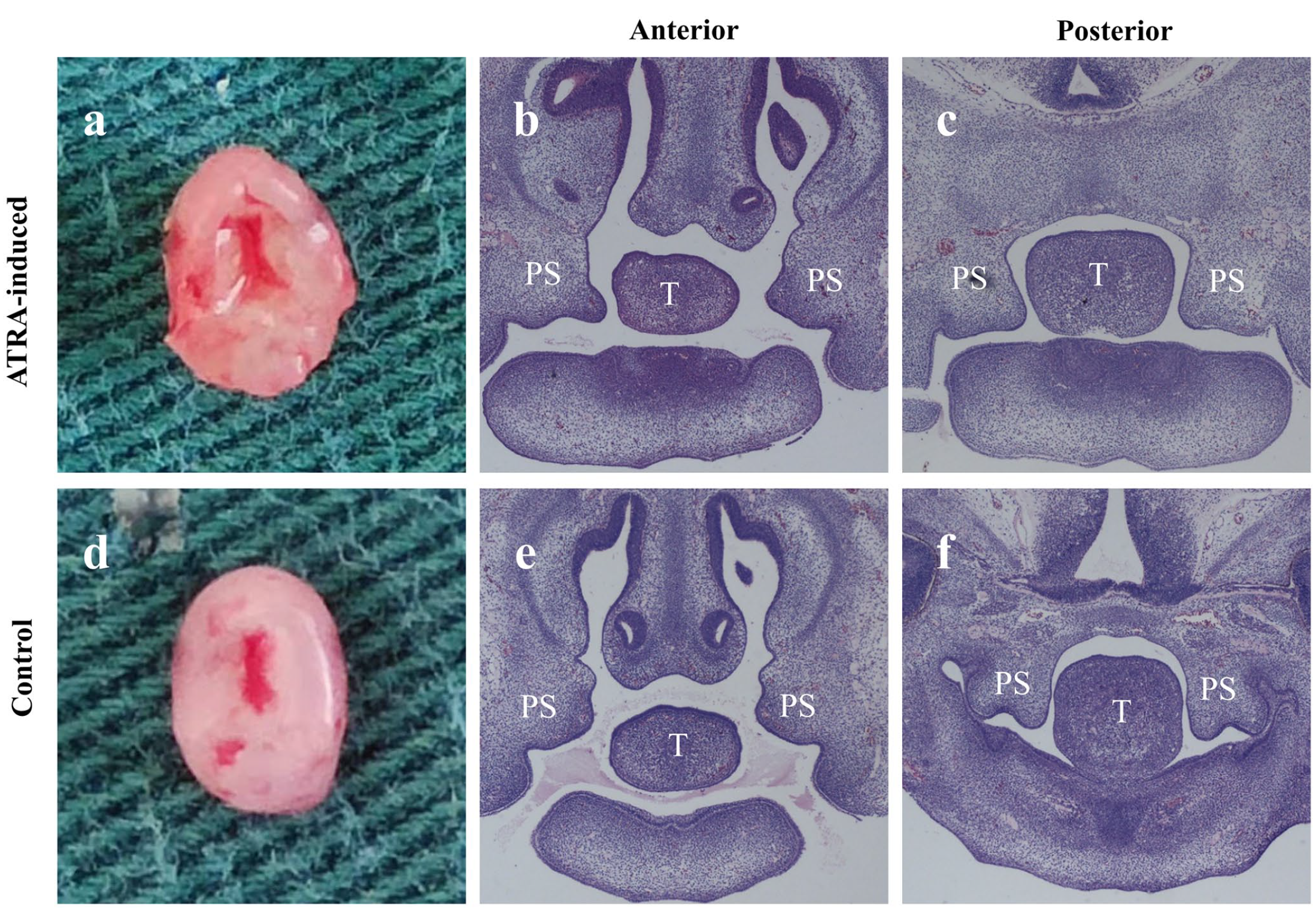
Gross morphology and histology (H&E) of palate shelf tissues at E13.5 in ATRA-induced versus control embryos. The palatal shelves were separated without fusion and grew vertically along the sides of the tongue in both groups. The width of the bilateral palatal shelf seam in ATRA-induced embryos was significantly larger than that in the control embryos (a, d). The bilateral PS elevation above the dorsum of the tongue and MEE fusion in the anterior and posterior domains was slightly delayed in ATRA-induced (b, c) versus control (e, f) embryos. (a, d) Gross morphology; (b, c, e, f) H&E staining results (×40). H&E: hematoxylin-eosin; PS: palatal shelf; T: tongue.
MiRNA and mRNA expression profiles
In total, results from RNA-seq analyses identified 1170 novel miRNAs, 97 of which were differentially expressed between the ATRA-induced and control groups, including 56 upregulated and 41 downregulated miRNAs in the ATRA-induced group. The heatmap in Figure 2(a), based on unsupervised hierarchical clustering analysis, visually represents the higher number of upregulated than downregulated miRNAs in the ATRA-induced group. A total of 1461 mRNAs were identified to be differentially expressed (P < 0.05, log2FC > 1) in the ATRA-induced group compared to the controls. The MA plot in Figure 2(b) shows a symmetrical distribution of the up- and downregulated mRNAs. The genes that were significantly differentially expressed are visualized as a volcano plot in Figure 2(c) and as a heatmap in Figure 2(d). Among the differentially expressed miRNAs and mRNAs, miRNA-470-5p and Fgfr1 were significantly upregulated and downregulated, respectively, in the ATRA-induced groups than in the controls.
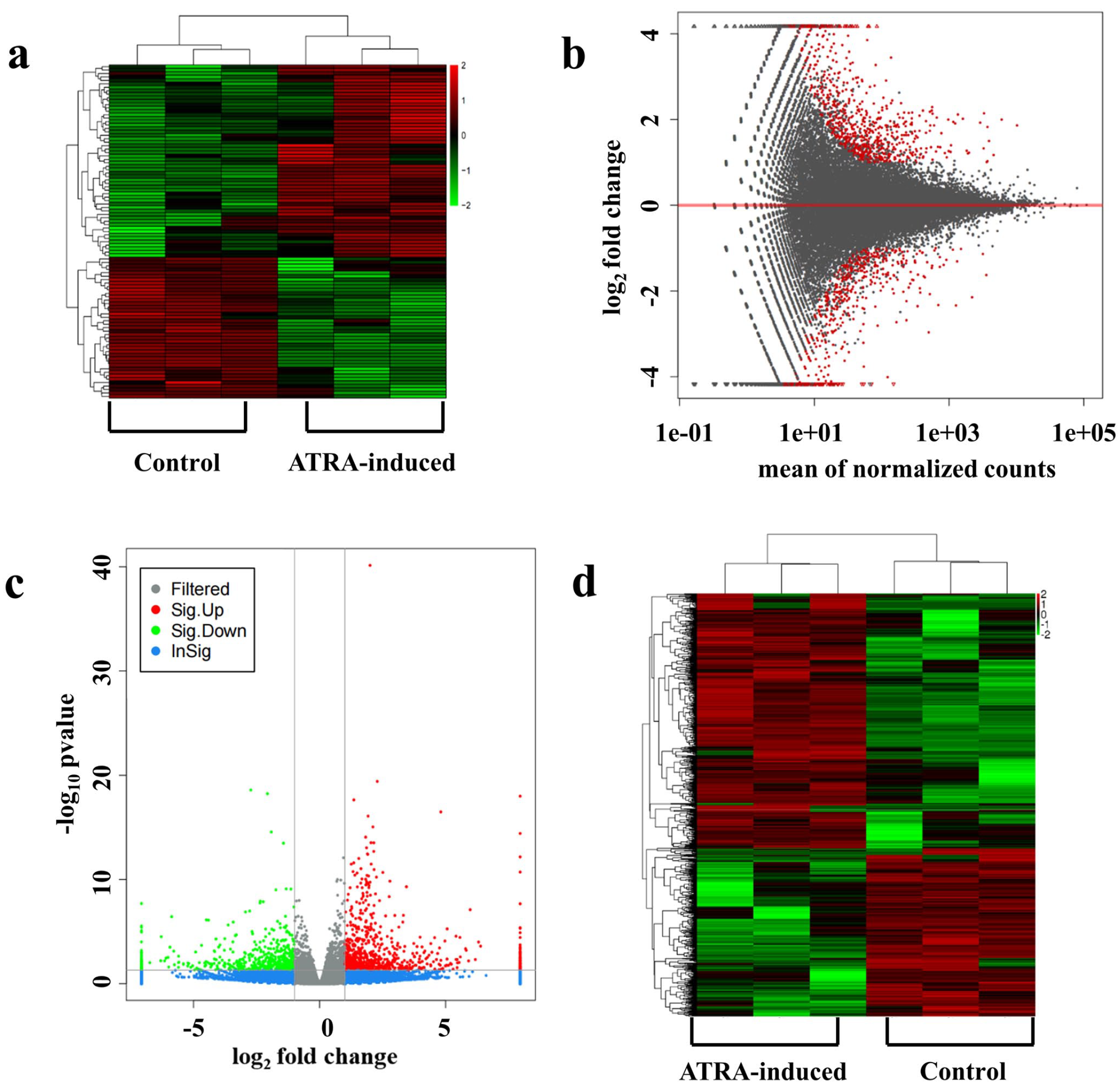
Heatmap showing results from unsupervised hierarchical clustering for differentially expressed miRNAs; red and green indicate high and low expression, respectively (a). MA plot for differential expression of mRNAs. The x-axis displays the mean of normalized counts from the expression of all samples, while the y-axis shows log2FoldChange. The red dots indicate genes exhibiting significantly different expression (b). Volcano plot showing the differentially expressed mRNAs in ATRA-induced versus control samples. Red and green dots indicate differentially expressed genes (DEGs). Gray and blue dots indicate non-DEGs. The y-axis shows log10 P value and the x-axis log2FoldChange (c). Heatmap indicating differences in mRNA expression profiles. Red and green indicate upregulated and downregulated mRNAs, respectively (d).
MiRNA-470-5p is predicted to directly target Fgfr1
To better understand the modulation mechanisms of miRNA-470-5p, potential miRNA-470-5p target genes were predicted through miRanda (Figure 3(a)) and TargetScan (Figure 3(b)). Results from both tools indicated that miRNA-470-5p has specific binding sites on the Fgfr1 sequence (Figure 3), suggesting that miRNA-470-5p might be a key factor regulating palatogenesis by targeting Fgfr1. Based on these results, this miRNA-mRNA pair was selected for further analyses.
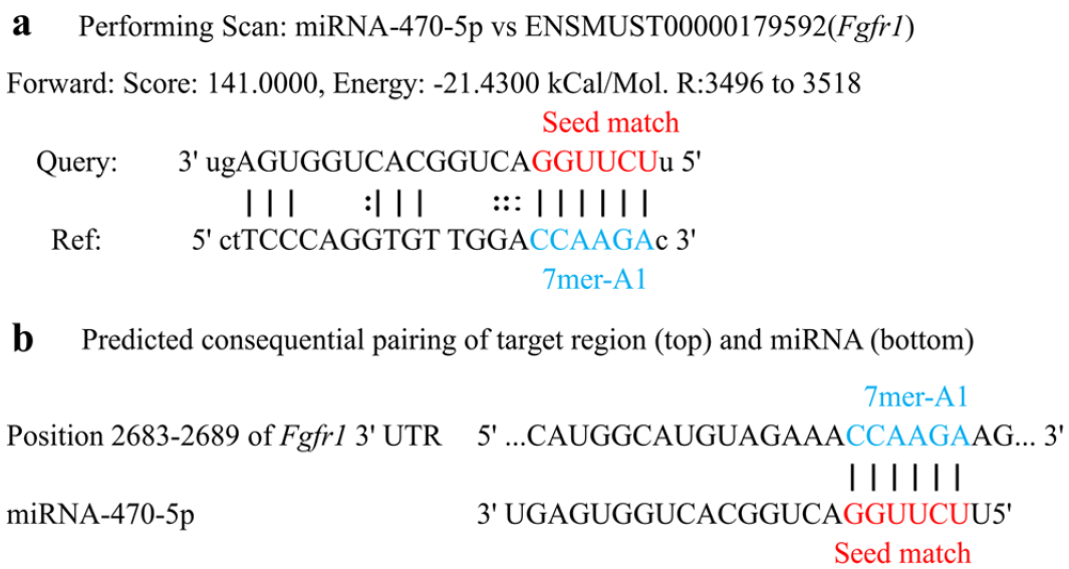
miR-470-5p versus Fgfr1-target binding sites predicted by miRanda software (a) and Target scan (b) in ATRA-induced and control samples. Site 7mer-A1: an exact match to positions 2–7 of the mature miRNA (the seed) followed by an “A.”
Validation of differential expression of miRNA-470-5p and Fgfr1 in the PS of the embryonic CP mouse model
RT-qPCR was conducted to validate miRNA-470-5p and Fgfr1 expression levels in embryonic PS tissues. Consistent with the RNA-seq results, the mRNA expression level of Fgfr1 was significantly downregulated (Figure 4(a)) and the miRNA-470-5p level was significantly upregulated (Figure 4(b)) in the ATRA-induced group compared with those of the control group.
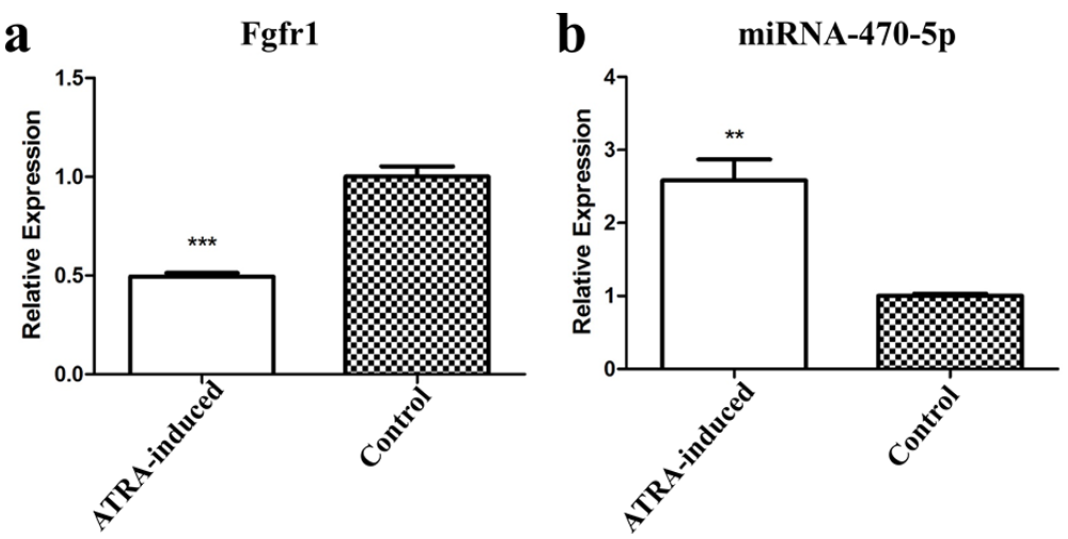
Relative gene expression levels of Fgfr1 (a) and miRNA-470-5p (b) in PS tissues in AT-RA-induced versus control groups. Gene expression was examined via RT-qPCR and normalized to that of the housekeeping gene GAPDH. **P < 0.01, ***P < 0.001.
Fgfr1 is a miRNA-470-5p target gene
To confirm the regulation effects between miRNA-470-5p and Fgfr1, luciferase reporter gene assays were conducted using plasmids encoding the Wt or Mut sequence of the Fgfr1 3′-UTR, with only the former containing the predicted miRNA-470-5p binding site. The sequences of Fgfr1-3′UTR-Mut and Fgfr1-3′UTR-Wt were, respectively, cloned to construct the reporter vectors. MiRNA-470-5p over-expression significantly reduced the luciferase activity of Fgfr1-Wt but had no effect on Fgfr1-Mut (Figure 5(a)). This result suggested that miRNA-470-5p acts directly on the target sequence of the Fgfr1 mRNA 3′UTR to suppress gene expression.
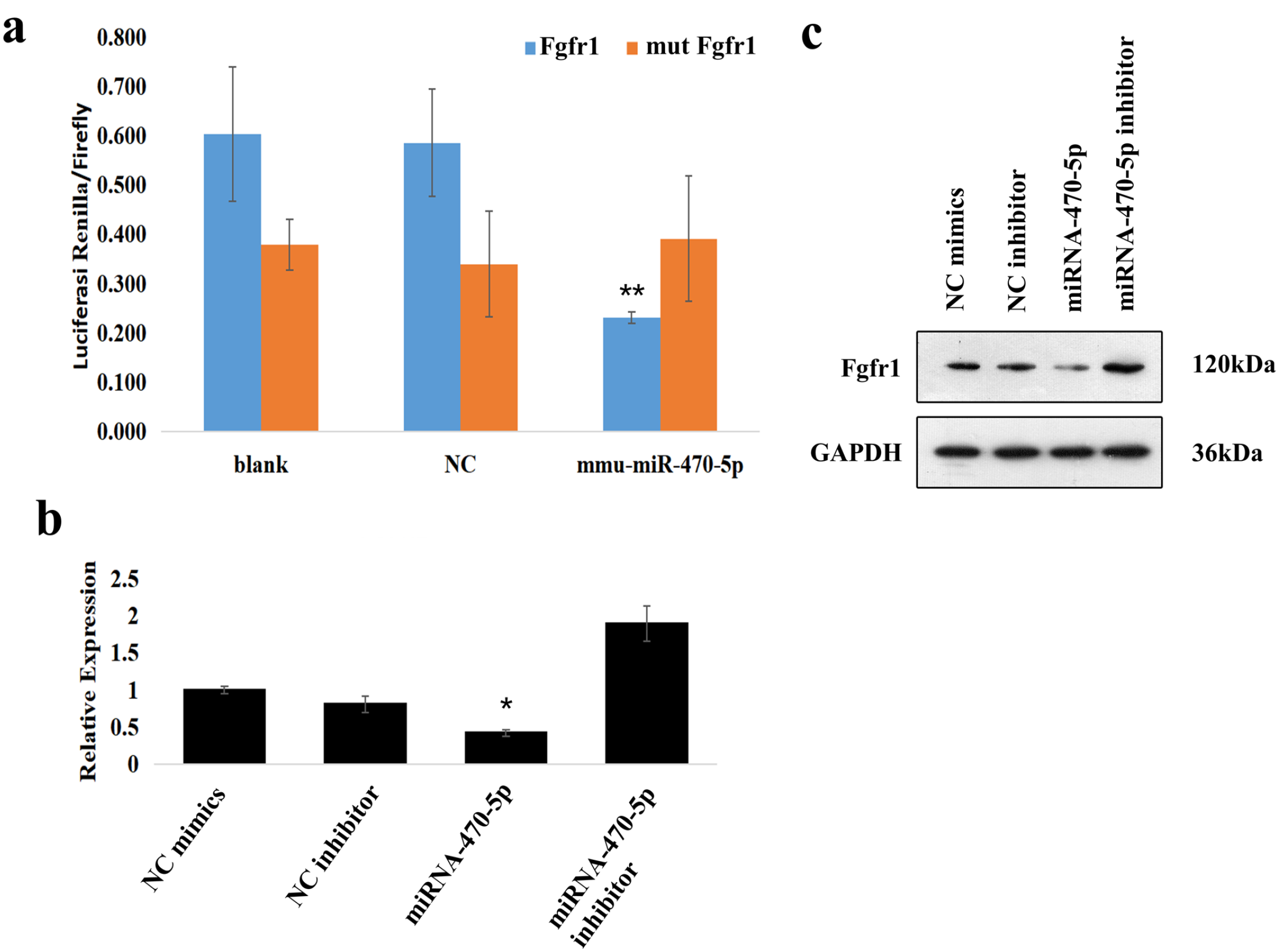
Luciferase reporter assays were performed in HEK293T cells. (a). Fgfr1 expression in mouse embryonic palatal shelf (MEPS) epithelial cells, using RT-qPCR (b) and Western blot (c) analyses. GAPDH was used as an internal control gene for the quantification of mRNA and protein expression. *P < 0.05 indicated significant over-expression of miRNA-470-5p and downregulation of Fgfr1 mRNA levels in miRNA-470-5p group compared to the other three groups. **P < 0.01 indicated that miRNA-470-5p significantly decreased the renilla/firefly luciferase activity of the Wt Fgfr1 3′-UTR construct.
To explore the potential mechanism by which Fgfr1 is regulated by miRNA-470-5p in CP formation, Fgfr1 mRNA and FGFR1 protein levels were analyzed in miRNA-470-5p silenced/over-expressing MEPS epithelial cells by RT-qPCR and Western blotting, respectively. miRNA-470-5p over-expression significantly inhibited FGFR1 expression at both the mRNA (Figure 5(b)) and protein (Figure 5(c)) levels. These results indicated that the Fgfr1 expression level is inversely related to the miRNA-470-5p expression level in CP tissues.
MiRNA-470-5p inhibited the proliferation of embryonic PS epithelial cells
To discover the possible biological function of miRNA-470-5p in CP formation, miRNA-470-5p-mimic, miRNA-470-5p-inhibitor, and the corresponding negative control oligonucleotides were, respectively, transfected into MEPS epithelial cells. The CCK-8 assay showed that over-expressing/suppressing miRNA-470-5p remarkably reduced/promoted the proliferation of MEPS epithelial cells at 24, 48, and 72 h (Figure 6(a)). Crystal violet staining showed that over-expressing/suppressing miRNA-470-5p appeared to reduce/promote the colony numbers of MEPS epithelial cells (Figure 6(b)). The calculated cell colony formation rates (colony numbers/initial seeded cells) further confirmed these trends (Figure 6(b)). Moreover, EdU staining revealed that over-expressing miRNA-470-5p significantly suppressed the DNA synthesis of the MEPS epithelial cells (Figure 6(c)). Collectively, these results indicated that miRNA-470-5p over-expression could inhibit the proliferation of MEPS epithelial cells and potentially lead to CP formation.
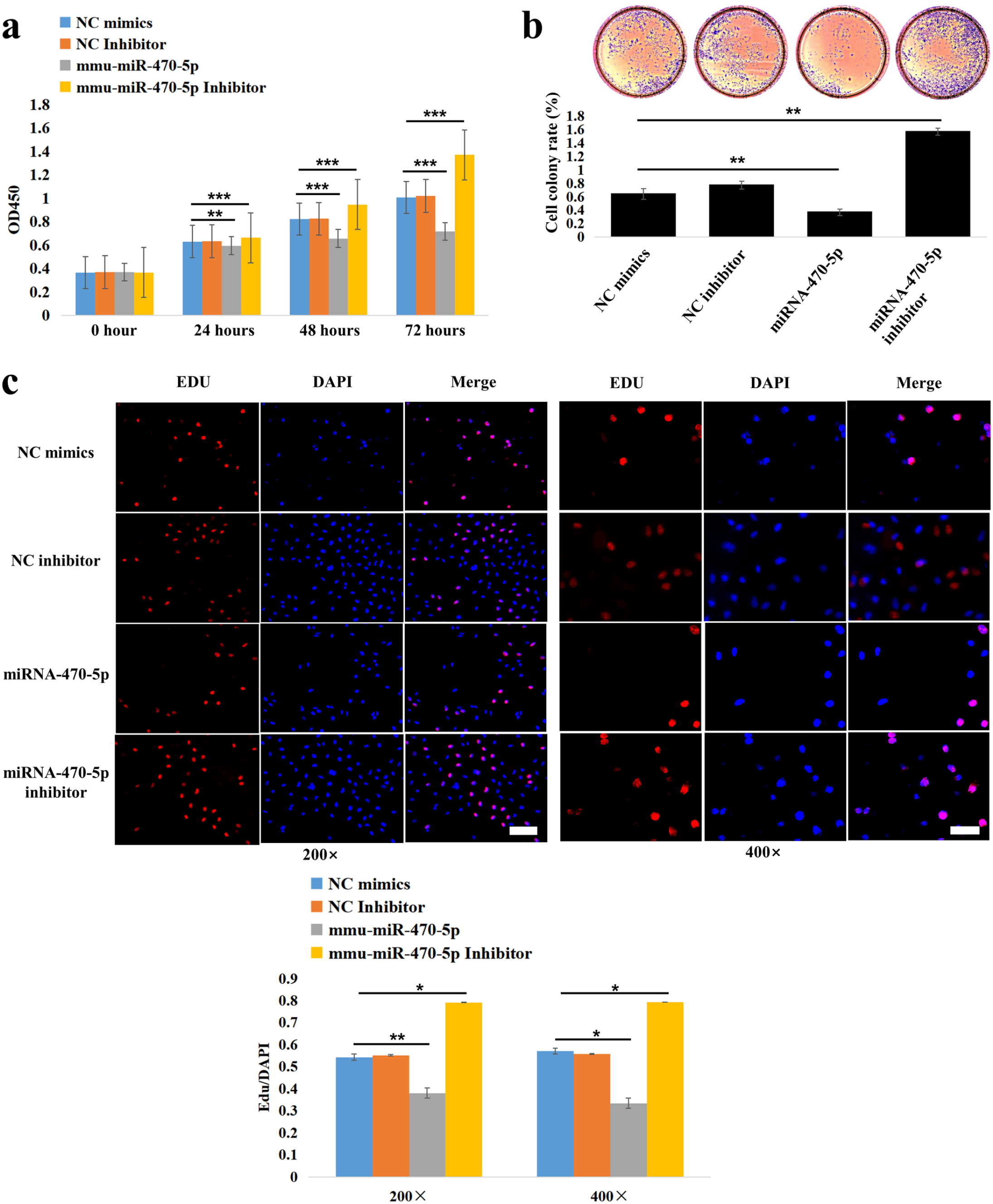
Inhibitory effect of miRNA-470-5p on the proliferation of MEPS epithelial cells was verified by CCK8 (a), colony formation (b), and EdU (c) assays. EdU-positive cells (red fluorescence). DAPI-stained cell nuclei (blue fluorescence). The comparison groups were miRNA-470-5p versus NC mimics, and miRNA-470-5p inhibitor versus NC mimics, respectively. *P < 0.05, **P < 0.01, ***P < 0.001. Scale bar (white): 100 µm (200×); 50 µm (400×).
Discussion
Accumulating evidence has confirmed that miRNAs play pivotal roles in palatogenesis.25,26 The significance of miRNAs in palate development has been widely studied through loss-of-function experiments of miRNAs; however, the detailed mechanisms remain unclear. We here provide the first evidence that miRNA-470-5p is potentially associated with embryonic palate development and CP pathology. Zhan et al. 27 found that miRNA-470-5p was involved in depressive behaviors and the outgrowth of hippocampal neurons in mice. However, the role of miRNA-470-5p during palatogenesis has not been reported until now. Moreover, the biological function of miRNA-470-5p and its molecular mechanisms underlying CP formation have not been reported. In this work, we found that miRNA-470-5p was highly upregulated, whereas its target gene Fgfr1 was significantly downregulated in CP tissues of an ATRA-induced embryonic mouse model. FGFR1 is a member of the fibroblast growth factor family, which has been shown to be a critical factor for palate development by mediating a variety of cellular responses. 28 Loss-of-function mutations in Fgfr1 were previously reported to delay cell proliferation in both mesenchymal and epithelial cells, thereby preventing palate shelf elevation. 29 Luciferase reporter assays identified that miRNA-470-5p acts directly on the target sequence of the Fgfr1 3′UTR to suppress Fgfr1 expression. Therefore, miRNA-470-5p may inhibit the EMT of MEPS epithelial cells by targeting Fgfr1 during palatogenesis. Indeed, we found that Fgfr1 expression was downregulated in the ATRA-induced group compared to the control group. Moreover, in vitro functional assays showed that miRNA-470-5p could suppress the cell proliferation and colony formation of MEPS epithelial cells. Thus, a possible relationship between palatogenesis and upregulation of miRNA-470-5p was demonstrated based on the following three lines of evidence: (1) miRNA-470-5p directly combines with the 3′-UTR of Fgfr1; (2) the RT-qPCR results showing the differential expression of miRNA-470-5p and Fgfr1 between the ATRA-induced group and control group were consistent with the RNA-seq results; and (3) gain- and loss-of-function experiments in vitro revealed that the upregulation of miRNA-470-5p could inhibit the EMT of MEPS epithelial cells by targeting Fgfr1.
The secondary palate of mammals is formed by bilateral shelves, which initially grow vertically arising from the maxillary process, and then begin to elevate and grow laterally on the tongue until they come into contact at the midline. 30 The transient MES is formed by adherence of the bilateral shelves during embryogenesis. Degradation of the MES leads to the failure of mesenchymal fusion, which may cause CP formation. Several studies have proven that the apoptosis of MEE cells, lateral migration of MEE cells, and EMT are three primary mechanisms by which secondary PS fusion is achieved. 31 EMT is a well-established process involved in cancer development, embryogenesis, and organ development, 32 and failure of the EMT step in palatogenesis has been shown to contribute to CP.19,31,33 Mice and humans share a high degree of similarity in their palatogenesis and genomes. 34 During normal palatogenesis, the PS, composed of an epithelium-covered mesenchyme core, grow down vertically along both sides of the tongue until E13.5 in mice. 35 MEE cells of the horizontal PSs contact, adhere, and fuse along the facial midline to form a continuous secondary palate from E14.5 to E17. Hence, the fate of MEE cells appears to be crucial for palate development. We chose the MEPS epithelial cells of mouse embryos as the object of study to investigate the etiological mechanism of CP. Erfani et al. 36 demonstrated that there is a key intershelf distance in the E13.5 mouse, beyond which the PS will not fuse in culture. In addition, they found that EMT was suppressed with physical separation and was nearly absent in the shelves that did not fuse. We therefore focused our in vivo study at E13.5, just prior to the period of rapid growth and elevation of the PS. Importantly, EMT is considered to be the key contributor to MES loss and subsequent mesenchymal fusion. 18
EMT is regulated through precise gene regulatory networks. Aside from the direct effects of gene expression, the change of RNA level can regulate EMT progression. 37 The differential splicing of RNA into mRNA has different functions in EMT, and the degradation of gene transcripts mediated by miRNAs determine the activity of key EMT proteins. Non-coding miRNAs that specifically bind to mRNAs can either promote the degradation or translation of mRNAs and thus regulate the EMT. 37 Our results indicated that miRNA-470-5p over-expression remarkably inhibits the proliferation of MEPS epithelial cells at E13.5. In other words, the quantity of cells transformed from MEPS epithelial cells to palatal mesenchymal cells is reduced upon over-expressing miRNA-470-5p. Overall, we concluded that the upregulation of miRNA-470-5p suppresses the EMT of MEPS epithelial cells during palatogenesis, leading to downregulated expression of its target gene Fgfr1, ultimately contributing to CP formation. The current study does have some limitations, such as a relatively small sample size and lack of gene knockout experiments. Future research should investigate these regulatory processes using in vitro organoid models38,39 and explore other signaling pathways that may be modulated by miRNA-470-5p.
Conclusions
The EMT process in CP is precisely modulated by numerous molecules, cytokines, and cellular signaling pathways. The present study identified, for the first time, that miRNA-470-5p is closely related to CP formation. Our results suggest that miRNA-470-5p could potentially suppress the EMT process through targeting the Fgfr1 3′UTR to facilitate the degradation of Fgfr1 mRNA. This study thus provides a theoretical foundation for miRNA-470-5p and Fgfr1 as a new class of biomarkers that could facilitate early screening and diagnosis, or the development of new strategies for the prevention and treatment of CP.
Footnotes
Acknowledgements
The authors thanked Shanghai Oe Biotech Co. Ltd (Shanghai, China) for the technical assistance. The authors would like to thank everyone involved in this study.
Authors’ Contributions
SS, MJZ, and YL contributed to the study idea and critical experimental design; JZ, MZ, MT, and YJ acquired, analyzed, and interpreted the data; JZ wrote the initial manuscript; SS and YL revised and approved the final review of the manuscript. All authors gave their final approval to the submitted version.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by Natural Science Foundation of Guangdong Province (grant number 2019A1515010346) and Cultivation of Medical Science Technological Talents and Clinical Technology Improvement Project of Shantou City (grant number 190917085269837).