
Abstract
Sickle cell disease (SCD) is an inherited hemoglobinopathy in which affected hemoglobin polymerizes under hypoxic conditions resulting in red cell distortion and chronic hemolytic anemia. SCD affects millions of people worldwide, primarily in Sub-Saharan Africa and the Indian subcontinent. Due to vaso-occlusion of sickled red cells within the microvasculature, SCD affects virtually every organ system and causes significant morbidity and early mortality. The neurological complications of SCD are particularly devastating and diverse, ranging from overt stroke to covert cerebral injury, including silent cerebral infarctions and blood vessel tortuosity. However, even individuals without evidence of neuroanatomical changes in brain imaging have evidence of cognitive deficits compared to matched healthy controls likely due to chronic cerebral hypoxemia and neuroinflammation. In this review, we first examined the biological contributors to SCD-related neurological complications and then discussed the equally important socioenvironmental contributors. We then discuss the evidence for neuroprotection from the two primary disease-modifying therapies, chronic monthly blood transfusions and hydroxyurea, and end with several experimental therapies designed to specifically target these complications.
Impact Statement
Sickle cell disease (SCD) is an inherited disorder that affects a predominately minority population in the United States and Europe, but millions of people in the developing world. Although more common than other inherited disorders, SCD continues to receive a disproportionately low percentage of research funding. In addition, although the life expectancy for those with SCD in the United States and Europe has improved over the last 20 years, the disease continues to have significant morbidity with virtually every organ system affected, particularly the brain. Individuals with SCD are subject to progressive cognitive decline beginning in infancy that directly impacts academic achievement, later job attainment, and quality of life. This minireview highlights the primary biological and environmental contributors to SCD-related neurological complications with the goal to emphasize SCD as a syndrome of accelerated aging and expand this topic to a wider audience outside of hematology.
Introduction
Sickle cell disease (SCD) affects over 100,000 people in the United States and millions worldwide.1,2 SCD includes a group of related hemoglobinopathies that result from a single base pair substitution at the sixth amino acid position of the beta globin gene, resulting in a change from a hydrophilic glutamic acid to a hydrophobic valine. This single substitution causes the mutated hemoglobin strand (known as hemoglobin S, HbS) to polymerize and to distort the red cell shape (“sickle”) under hypoxic conditions. The hallmark complication of SCD is pain due to aggregation of sickled red cells within the microvasculature of the long bones, resulting in ischemia and infarction. However, virtually every organ system can be affected, leading to significant morbidity and early mortality.3,4 The genotypes HbSS and HbS-0β thalassemia, together known as sickle cell anemia or SCA, are generally the most severe forms and are associated with significant neurological sequelae.
The neurological complications of SCA are particularly devastating and include overt stroke, silent cerebral infarctions (SCIs), and progressive cognitive decline with or without evidence of cerebral injury.5,6 Indeed, prior to the advent of disease-modifying therapy and transcranial Doppler (TCD) screening for primary stroke prevention, 10% of children with SCA were expected to have an ischemic stroke by age 20 years. 7 In addition, close to 40% of children have evidence of SCI by age 14 years.8,9 SCIs are defined as one or more areas of hyperintensity on magnetic resonance imaging (MRI) and are visible on two or more planes on T2-weighted and fluid-attenuated inversion recovery (FLAIR) sequences, without an associated clinical sign or symptom. The prevalence of SCI increases to over 50–60% among adults with SCA.10,11 However, even children and adults without evidence of MRI changes score on average 10 points lower on measures of full-scale intelligence (FSIQ) with evidence of cognitive impairment compared to unaffected peers.5,6
These cognitive changes begin early, with infants as young as nine months of age already scoring lower than expected on measures of infant development and are progressive with a loss of one FSIQ point every two years by adolescence.12,13 Not only are the array of SCA-related neurological complications complex and variable, the etiologies of these complications are multifactorial, with contributions from both disease-related mechanisms and psychosocial factors (Figure 1 and Table 1).14–17 In this review, we first provide a summary of neurological complications that primarily affect those with the more severe SCA genotypes and then explore the contributors to the pathophysiology of cognitive decline in SCA and discuss tentative therapies. Finally, we give a brief overview of the less common and generally more mild neurological complications and disease-related cognitive deficits in the non-SCA genotypes, specifically HbSC and HbS-β+ thalassemia.
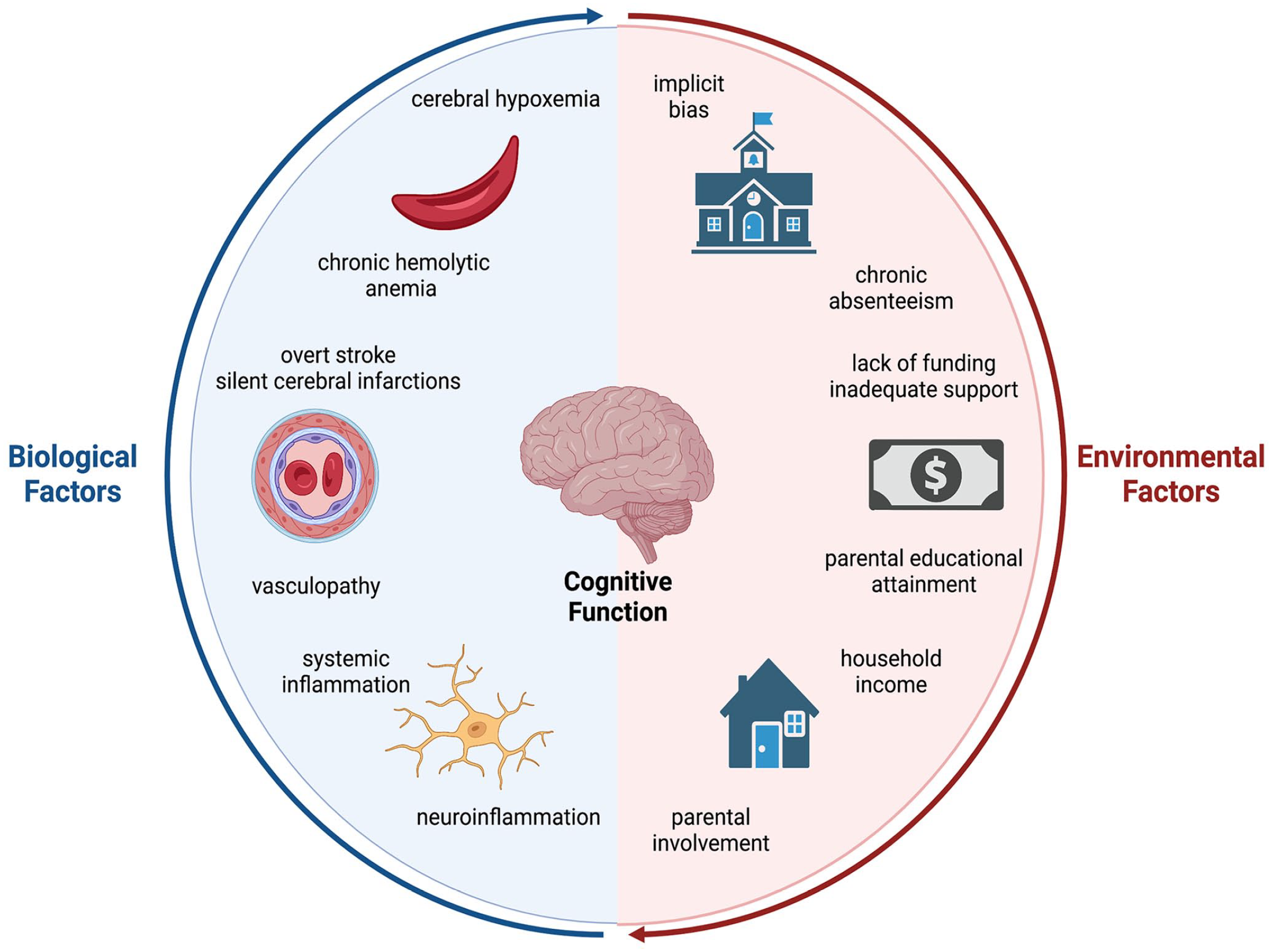
The biological and environmental contributors to cognitive decline in sickle cell disease. Cognitive decline in individuals with sickle cell disease (SCD) is complex with diverse biological and environmental contributors. The biological components include chronic hemolytic anemia, cerebral hypoxemia, vasculopathy, including overt stroke and silent cerebral infarctions, and systemic and neuroinflammation. Societal and environmental components span from childhood household income, parental educational attainment, and chronic school absenteeism to a lack of funding for SCD research and societal implicit bias. As illustrated through the circular model, all of these contributors are interconnected and influence one another.
Summary of the pivotal studies in SCD-related cognitive decline and prevention.
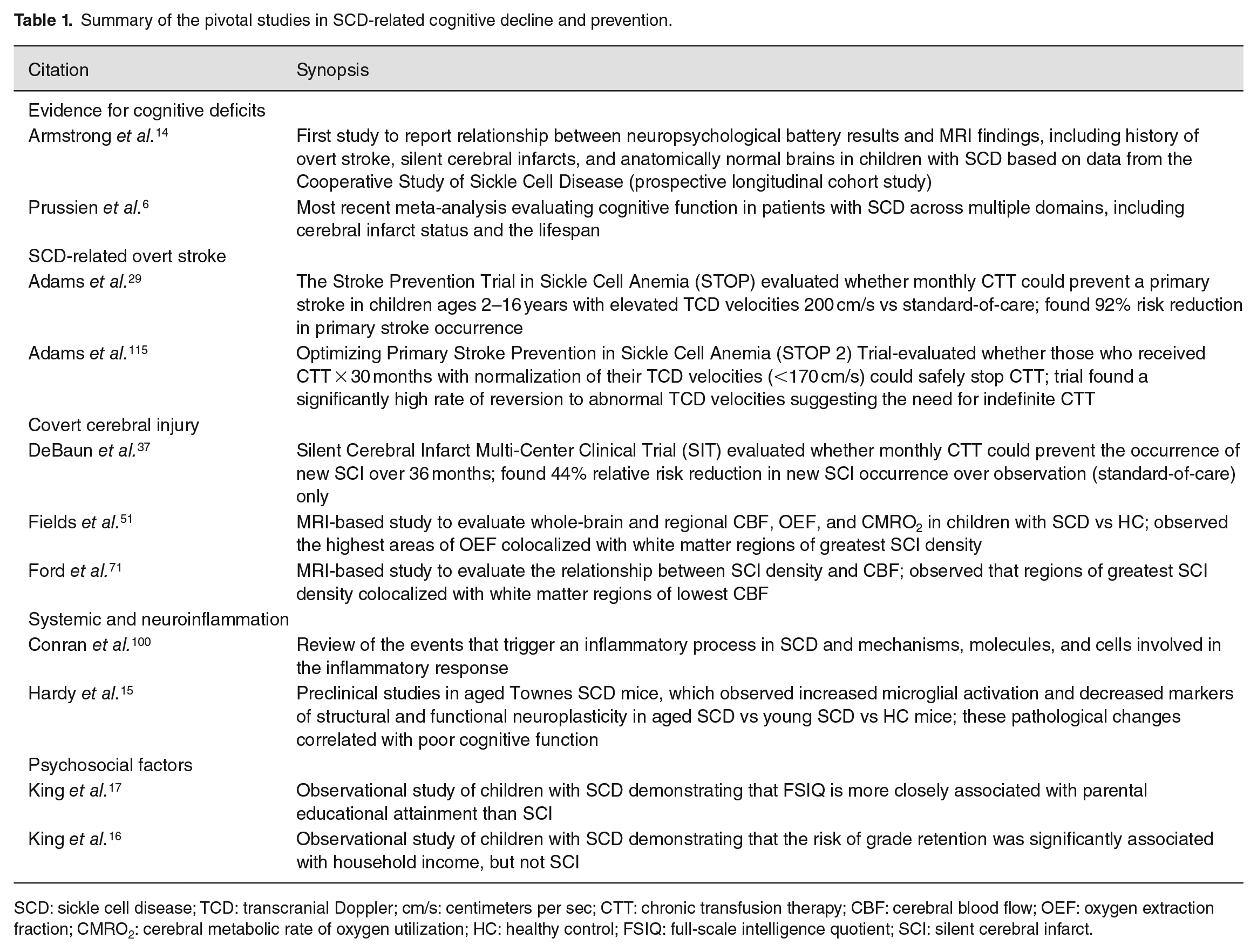
SCD: sickle cell disease; TCD: transcranial Doppler; cm/s: centimeters per sec; CTT: chronic transfusion therapy; CBF: cerebral blood flow; OEF: oxygen extraction fraction; CMRO2: cerebral metabolic rate of oxygen utilization; HC: healthy control; FSIQ: full-scale intelligence quotient; SCI: silent cerebral infarct.
Overt cerebrovascular injury
Without disease-modifying therapy, individuals with the SCA genotypes are at increased risk for both ischemic and hemorrhagic stroke across the lifetime compared to unaffected populations. Ischemic stroke demonstrates a bimodal age distribution with a first peak in childhood at age four to five years, typically due to vaso-occlusion of the large vessels of the Circle of Willis (true SCA pathology) and again in the fifth decade, likely related to the development of stroke risk factors (such as hypertension and atherosclerosis, independent of SCA) among adults with SCA, similar to those of the general population. 7 The incidence of hemorrhagic stroke peaks in the third decade for still unclear reasons. 7 Not surprisingly, individuals with a history of large vessel ischemic stroke, particularly in childhood, also develop significant cognitive deficits with an average FSIQ = 74, well below the normative mean of 100, on a standard neuropsychological battery, as identified by the most recent meta-analysis of cognitive function in SCA. 6 The presence of overt, typically large vessel stroke is associated with a large effect size across all domains, including FSIQ, verbal reasoning, perceptual reasoning, executive function, and processing speed (Hedges’ g = −1.35 to −1.82). 6 This observation is different from individuals without SCA who experience a stroke and who tend to have deficits localized to the region of infarct. There may be several reasons for this. Preclinical studies show that SCA mice have a higher sensitivity to hypoxia and that a large vessel occlusion results in a larger infarct area compared to AA mice.18–20 The presence of anemia also results in altered hemodynamics (discussed in detail further in this article) and widespread chronic hypoxia, and hypercoagulability. The compensatory mechanisms are maximally taxed, and a large supply-demand mismatch such as a stroke can therefore affect regions with a precarious oxygen supply to demand ratio and result in deficits via direct structural injury, thrombosis, and inflammation farther from the region of infarct.
SCA-related cerebral vasculopathy occurs most commonly in the anterior cerebral, middle cerebral, and distal internal carotid arteries with a prevalence of ~10% on magnetic resonance angiography (MRA) in children with SCA.21,22 This vasculopathy is a spectrum, ranging from moderate stenosis (50–75% vessel stenosis) to severe stenosis (75–99%) and occlusion to finally the development of SCA-related moyamoya syndrome, in which collateral vessels form with the characteristic “puff of smoke” appearance on arterial imaging.23–25 These collaterals are progressively less efficient at providing alternative blood flow until the affected individual ultimately becomes dependent on external carotid-derived blood flow for cerebral perfusion. 24 Adams et al.26–28 showed that not only does TCD ultrasound correlate strongly with the presence of stenosis in these vessels, but that elevated TCD ultrasound velocity in any of the above-mentioned arteries (distal internal carotid artery [ICA], middle cerebral artery [MCA] or anterior cerebral artery [ACA]), defined as non-imaging TCD velocities ⩾ 200 centimeters per second (cm/s) (also known as the time-averaged maximum mean cerebral blood velocity, or TAMMV), is associated with a 40 times higher risk for stroke compared with normal velocity (or non-imaging TCD velocity < 170 cm/s). The Stroke Prevention Trial in Sickle Cell Anemia (STOP) showed that regular monthly blood transfusions are able to normalize TCD velocity and effectively lower the risk for stroke by 70%. 29
More recently, several studies have noted a relationship between extracranial internal carotid artery stenosis/occlusion and stroke risk, even in the absence of intracranial arterial abnormalities.30–32 In addition, extracranial internal carotid artery stenosis is a major cause of ischemic stroke in adults without SCA.33,34 In a study of more than 400 children with SCA, Bernaudin et al. 9 found that a TAMMV velocity of at least 160 cm/s on Doppler ultrasound in the extracranial internal carotid correlated with MRA-confirmed stenosis. A limitation of this study was its cross-sectional nature; thus, whether this elevated velocity is predictive of an increased stroke risk is unknown. In addition, to date, no randomized trials have evaluated whether chronic blood transfusion therapy prevents SCA-related stroke secondary to extracranial internal carotid artery stenosis/occlusion. Thus, neither the 2014 National Heart, Lung, and Blood Institute (NHLBI) Guidelines for the Management of SCD nor the 2020 American Society of Hematology (ASH) SCD-related Cerebrovascular Disease Guidelines include extracranial internal carotid artery screening in their TCD recommendations.36,37
Although the mechanisms for the development of large vessel stenosis remain unclear, the abnormal physiological properties of the sickled red cells and their aberrant interactions with the endothelium, as they navigate through the Circle of Willis, ICA and its branches, and cerebral microcirculation,38,39 are postulated to contribute. Specifically, evidence from histological studies showed significant intimal hyperplasia with fibroblast and smooth muscle proliferation and focal medial fibrosis.40,41 Thus, sickled cells are postulated to become adherent to the blood vessel wall and are subsequently forcibly removed by turbulent blood flow; 42 this causes local intimal injury and secondary platelet aggregation and growth factor release, including platelet-derived growth factor and transforming growth factor, two potent stimulants of fibroblast, and smooth muscle formation.43,44 The intimal hyperplasia results in flow turbulence, which leads to abnormal (supraphysiological or subphysiological) wall shear stress 39 which further perpetuates the cycle of endothelial injury and overcompensation.42–44
The intrinsic properties of the sickled red cells (blood cell rheology) are also thought to contribute. Normal red blood cells (RBCs) can compensate for the changes in blood flow by extensive passive shape changes in the setting of high shear rates through arteries and aggregate formation (“Rouleaux forms”) under low shear through veins. Individuals with SCA, by contrast, have higher blood viscosity at baseline than unaffected individuals, as well as worse RBC deformability and decreased aggregation; these characteristics become more pronounced in the deoxygenated state and make the RBCs more subject to hemolysis.45,46 Hemolyzed RBCs release hemoglobin, which combines with nitric oxide, a key regulator of vascular and endothelial health, to form methemoglobin and nitrate. In addition, heme and heme-iron activate reactive oxygen species, which further scavenge nitric oxide, 46 while the lysis of sickled RBCs releases arginase, which in turn destroys arginine, the substrate for nitric oxide production. 47 This lack of nitric oxide, a potent vasodilator, results in endothelial dysfunction, impaired vasomotor tone, and vessel vasoconstriction, which further perpetuates the cycle of RBC vessel wall adhesion and injury and resultant intimal hyperplasia and fibrosis.42,45,46 Two studies using computational fluid dynamics to model blood flow in the distal ICA and its main branches have predicted areas of increased blood flow velocity and abnormal wall shear stress. Both studies reconstructed the distal ICA and its main branches from MRA images obtained from patients with SCA.39,48 Although generalizability is limited given the small sample size, these abnormal changes were not seen in reconstructions derived from a healthy control. 48
Covert cerebrovascular injury
Although overt cerebral injury can be devastating, a larger percentage of individuals with SCA have evidence of covert neurological injury, which predispose to cognitive deficits. 37 SCA-related covert cerebral injury includes SCIs, blood vessel tortuosity, lacunar infarcts, and chronic hypoperfusion injury.39,49–52
SCIs
SCIs are areas of white matter hyperintensity at least 3 mm in the largest dimension, seen on at least two views on FLAIR and/or T2-weighted MRI and are not associated with a focal neurological sign or symptom. 50 SCIs are most commonly bilateral, multiple, and located within the frontal and parietal deep white matter, followed by the basal ganglia and thalamus; other areas commonly involved include the parietotemporal deep white matter, frontal cortex, and parietooccipital cortex. 53 Data from the Cooperative Study of Sickle Cell Disease (CSSCD), the largest longitudinal cohort study of SCD to date, showed that individuals with SCI scored significantly lower on multiple subsections of a standard neuropsychological battery, including both verbal intelligence and FSIQ. 54 This suggests that while SCIs are not associated with a focal neurological sign or symptom, they are not particularly silent in that they are associated with cognitive deficits/impairment. This is unsurprising, given that the areas most commonly affected by SCI are also those involved in executive functioning and attention. 55 The most recent meta-analysis of cognitive function in SCD found medium effect from the presence of SCI in executive function (Hedges’ g = −0.52) and large effect across all other domains, including FSIQ, verbal reasoning, and processing speed (g = −0.97 to −1.03) with the average FSIQ = 85, a full standard deviation away from the normative mean of 100, further demonstrating that these “silent” infarctions are not truly silent. 6 Furthermore, even individuals with SCA without MRI detectable evidence of cerebral injury have an average FSIQ = 92, half a standard deviation below the normative mean; the diagnosis of SCD conferred medium effect (g = −0.54 to −0.65) across all cognitive domains evaluated by Prussien et al. 6
Blood vessel tortuosity
Neuroimaging data have shown that individuals with SCA have tortuous arteries and arterioles, as well as abnormal venule architecture.14,30,39,49,56,57 Rodent models of SCA also corroborate the presence of abnormal topology, with a tortuous capillary and cerebral microvascular network, increased RBC flux velocities, and shorter microvascular branch length. 52 While the exact pathophysiology of increased tortuosity is unknown, it can alter blood flow and increase the risk of vaso-occlusion and tissue hypoxemia. Therefore, not surprisingly, increased tortuosity has been linked to cognitive impairment. 58
Cerebral microinfarcts
Cerebral microinfarcts are defined as small, microscopic areas of tissue necrosis due to ischemia that are reliably visible on histological examination and newer, more powerful seven Tesla (7T) MRI. 59 Despite their size, microinfarcts can be widespread throughout the brain and can result in cognitive dysfunction by affecting regions remote from the lesion. 60
Clinical studies in non-SCA populations with ischemic heart disease have established a robust link between the burden of cerebral microinfarcts and cognitive impairment.61–64 Preclinical studies in non-SCA rodent models provide evidence that a single cerebral infarct can affect a region 12 times the original volume and result in impaired learning and memory despite only affecting a small tissue area.65,66 The hemodynamic changes from a microinfarct can also persist for weeks. Mechanistic studies show that microinfarcts can result in dendritic spine loss, neuronal death, white matter changes, blood–brain barrier leakage, persistent activation of microglia and astrocytes, and impaired glymphatic drainage which disrupt neuronal connections and can affect learning, memory, and executive functioning.60,65,67 In addition, microinfarcts have been documented in preclinical models of SCA particularly in the cortical regions. 52 A recent study showed that SCA mice had a 2.5 times higher rate of spontaneous cerebral microinfarcts compared to age and sex-matched control mice. 52 SCA mice also have significantly larger microinfarcts compared to controls.38,52 While the underlying mechanism for cerebral microinfarcts is not yet known, the available evidence suggest that it could be a result of occlusion of cortical microvessels (arterioles, capillaries, and/or venules).38,52
Although their presence is not yet confirmed in humans in SCA, based on preclinical data and translation from other human disease states, it is plausible to conclude that individuals with SCA are also likely to have microinfarcts. Similar to inferences from animal studies, the etiology of cortical microinfarcts in humans is still unclear; however, available evidence suggests that it could be from a combination of cerebral large and/or small vessel disease, microthrombi, and hypoperfusion. 60 All such processes are certainly seen in SCA with chronic anemia leading to hypoperfusion, a heightened prothrombotic state, and the presence of cerebral macro and microvasculopathy.
Hypoperfusion and anemia
Despite its size, the brain has one of the highest rates of oxygen consumption compared to the remainder of the organ systems and is uniquely vulnerable to hypoxemia. 68 To maintain a steady cerebral metabolic rate of oxygen utilization (CMRO2), the brain relies on autoregulatory mechanisms that alter arteriolar diameter to maintain cerebral blood flow (CBF). 69 The brain also maintains the cerebral metabolic rate by increasing the percentage of oxygen extracted from the blood (OEF, oxygen extraction fraction). Thus, CMRO2 is regulated by OEF, CBF, and the arterial oxygen content of the blood (CaO2).51,70,71
In SCA, the combination of a lower baseline hemoglobin from increased hemolysis and RBC turnover and the lower oxygen affinity of HbS versus hemoglobin A (HbA) results in a lower cerebral oxygen saturation compared to even healthy controls with anemia from other causes.72,73 Preclinical studies also show that compared to age-matched controls, SCA mice have lower brain tissue oxygenation evidenced as an increased expression of hypoxia signaling pathways. 19 Interestingly, rodent models of SCA seem to show that low cerebral oxygen saturation may precede neuronal injury and cognitive dysfunction. 74
To normalize oxygen delivery and maintain CMRO2 in SCA, there is a compensatory increase in CBF. 75 In support of this, preclinical studies demonstrate increased velocities in cortical vessels which correlate with cognitive deficits pertaining to learning and memory.38,52 Clinical studies also support the presence of increased baseline velocities and their association with poor cognitive outcomes.51,76–79 Furthermore, CBF is not increased globally and appears to be decreased in deep white matter.51,80 While the exact mechanism for this phenomenon is unknown, this has an important implication, as the capillary density is often the lowest in deep white matter,81,82 and failure to increase CBF to maximize oxygen delivery in the setting of anemia makes this region particularly vulnerable to ischemia. 80 Indeed, it is not surprising that most SCIs are often located in deep white matter that appears to have the slowest CBF. 71
In addition to increasing CBF, increasing OEF may be another important mechanism to compensate for decreased blood oxygen content.51,83,84 There is controversy whether OEF is globally increased or decreased in individuals with SCD.83,85,86 Furthermore, the exact mechanisms of an increase or a decrease in OEF are not known. OEF changes may correlate to a region’s metabolic need, capillary density, and blood flow and overall hematocrit.87–89 Regardless, high OEF has been associated with poor cognitive outcomes, specifically those that pertain to executive functioning. 76 Interestingly, areas of high OEF correlate with the areas of highest density of SCI, suggesting increased vulnerability to ischemia and infarction. 51 Given the decreased CBF elevation in response to anemia in deep white matter, an increase in OEF may not be sufficient for any additional metabolic demand, thus increasing the risk of hypoxia and infarction. 51 The work of Kwiatkowski et al. 53 further clarified that SCIs most commonly occur bilaterally in the frontal and parietal deep white matter followed by the basal ganglia and thalamus. This is further supported by a recent diffusion-weighted imaging study in adults with SCA, which showed normal-appearing deep white matter regions with impaired microstructure that extend beyond the areas of the highest infarct density. 90 In addition, the areas most affected by SCIs are also those most involved in executive function and attention. 55
Neuroinflammation
Although a role for neuroinflammation in cognitive decline has not yet been documented in humans with SCA, neuroinflammation through primarily activated microglia (the resident immune cells of the central nervous system [CNS]) has been associated with cognitive decline across neurodegenerative disorders in humans, including in Alzheimer’s disease, frontotemporal dementia, and HIV-related cognitive decline.91–96 Recent preclinical studies from sickle cell mouse models suggests that neuroinflammation is likely to play a significant role in the pathobiology of SCA-related cognitive decline.15,97 Studies in individuals with Parkinson’s disease show the presence of microglial activation prior to the onset of dementia with the degree of microglial activation correlating with performance on cognitive measures. Therefore, microglial activation may predate and play a major role in progression of dementia in Parkinson’s disease. 98 Similar studies done in Alzheimer’s as well as supranuclear palsy also show microglial activation prior to the onset of significant cognitive decline suggesting that microglial activation could be playing a role in disease progression and associated cognitive decline. 99
SCD is fundamentally a proinflammatory disorder. 100 Studies have shown a positive correlation between systemic inflammation and neuroinflammation;101–104 proinflammatory cytokines released as a result of a systemic inflammatory response may influence the microenvironment in the brain by activating microglia and astrocytes.104–106 Peripheral leukocytes may also cross the blood–brain barrier and infiltrate the brain parenchyma and initiate or accentuate the neuroinflammatory process. 107 The subsequent changes can result in decreased maturation of the neuronal progenitor cells, increased elaboration of cytokines by microglia, and further recruitment of peripheral leukocytes. These cellular changes directly impact cognition.97,108 In one mouse model of SCA, the authors demonstrated loss of neuronal density and shrinkage of neurons in the cerebellum and hippocampus, which was associated with poor performance on learning and memory function, as assessed by the Morris water Maze and T maze test, as well as increased anxiety/depression-like behaviors assessed by the open field test. 107 In addition, Hardy et al. 15 observed increased microglial activation, decreased dendrite arbors and dendritic spine density (markers of structural neuroplasticity), increased proportion of immature dendritic spines (a marker of functional neuroplasticity), and increased density of peripherally derived CD45+ mononuclear cells in the hippocampus of aged Townes humanized SCA mice; all of these pathological changes correlated with poor cognitive function compared to younger SCA and healthy control mice. Both the behavioral changes and histological changes were abrogated by treatment with minocycline, a tetracycline antibiotic with antineuroinflammatory activities that effectively crosses the blood–brain barrier. 15
Psychosocial factors
As with many complex disease processes, the role of environment cannot be ignored in the development of SCA-related cognitive deficits, particularly in childhood. Individuals with SCD are disproportionally affected by social determinants of health, 109 which have been linked to poor health outcomes across multiple disease states.110–112 Regarding specifically SCA-related cognitive deficits, King et al. 17 found that FSIQ in school-aged children with SCA was more closely associated with parental educational attainment (head of household with no more than high school vs some college education) than the presence of silent cerebral infarcts in the affected child. In addition, the risk for grade retention in children with SCA was not associated with silent infarcts, but significantly associated with household income; children in the lowest quartile for household income had an odds ratio of 6.4 for retention compared to those of the highest quartile. 16 Not surprisingly, the influence of psychosocial factors on cognitive function also begins early; parental education and household income were associated with performance on measures of infant development in children 7–18 months old. 12 Furthermore, formal measures of the living environment using the HOME questionnaire, which includes observations of the child–parent interaction and quantity/quality of learning stimuli available in the home, moderately correlated with measures of infant development in a cohort of children ages 2–42 months old. 113 Finally, Fields et al. 114 demonstrated the ability to mitigate psychosocial contributions to cognition with early intervention; in a home visitation program for children ages 3–36 months with all SCD genotypes and their parents that focused on strengthening the parent–child relationship and knowledge/language acquisition, those enrolled in the program had a significant improvement in the cognition and expressive language subscales on the Bayley Scales of Infant/Toddler Development-III, a primary developmental assessment tool. In contrast, those who were followed longitudinally, but were not involved in the program, had no improvements and actually a trend toward worse performance on the cognition, gross motor, and fine motor subscales. 114
Therapies
Unfortunately, currently no specific therapies exist for treating and/or ameliorating the impact of cognitive deficits in individuals with SCA. However, chronic red cell transfusions have long been known to reduce stroke risk,29,115,116 while early evidence suggests that the other primary disease-modifying therapy hydroxyurea may also be efficacious, particularly in young children.117–119 Here, we provide the evidence for both disease-modifying therapies and their known limitations, as well as briefly touch on interventions still under investigation (Table 2).
Current SCD disease-modifying therapies and experimental cognition-specific targets.

SCD: sickle cell disease; NMDA: N-methyl-
Chronic transfusion therapy
As mentioned earlier, the STOP trial showed that routine monthly blood transfusions can normalize elevated TCD velocities and effectively lower the risk for stroke by 70% in children with SCA who were at high risk for stroke, evidenced by an elevated TAMMV TCD velocity (⩾200 cm/s). 29 The Optimizing Primary Stroke Prevention in Sickle Cell Anemia (STOP 2) Trial showed that cessation of chronic transfusion therapy (CTT) after sustained normalization of TCD velocities can unfortunately cause recurrence of the elevated TCD velocity, even in patients without evidence of severe large vessel stenosis on MRA. 115 In addition, CTT significantly reduces the risk for a second ischemic stroke in children with SCA with a prior stroke. 120 Thus, based on these data, the current NHLBI Sickle Cell Disease Guidelines recommend indefinite CTT as secondary stroke prophylaxis for both adults and children with a history of stroke to minimize the risk of stroke recurrence. 36
Chronic blood transfusions have also been shown to be protective against covert SCD-related cerebral disease. The Controlled Trial of Transfusions for Silent Cerebral Infarcts in Sickle Cell Anemia (SIT Trial) found a 58% relative risk reduction in the development of new or progression of existing SCIs in children randomized to receive monthly CTT vs observation only over a period of three years. 50 Hood et al. 116 observed that children on chronic monthly transfusions scored significantly better on a brief cognition battery shortly after receiving their monthly transfusion compared to long after their CTT (i.e. immediately prior to their next transfusion); on subsequent analysis, change in total hemoglobin concentration, but not percentages of HbA or HbS, was associated with improvement in performance on cognitive testing, suggesting that the observed benefits might be in part due to an increase in oxygen-carrying capacity and decreased strain on the cerebral metabolic rate. Also supporting this hypothesis is the observation that chronic transfusions decreased both global and regional CBF and OEF while maintaining the cerebral metabolic rate in a cohort of patients with SCA on CTT for primary stroke prevention, suggesting a decrease in cerebral metabolic stress and therefore lower risk for ischemia, particularly in the most vulnerable areas of the deep white matter. 84
However, chronic transfusions are not without significant side effects, particularly the risk for alloimmunization from repeated exposure to minor RBC antigens. Although the latter can be minimized by extended RBC matching, alloimmunization may still occur and can make locating and matching blood difficult. 121 The other primary risk is for progressive hepatic iron overload. Thus, individuals with SCA on CTT require routine screening and often initiation of chelation or phlebotomy programs. The use of erythrocytapheresis (RBC exchange transfusion) can reduce the risk for iron overload, but requires placement of implanted central lines, which increase the risk for venous thromboembolism in a population already at heightened risk for potentially life-threatening thrombosis. 122
Hydroxyurea
Hydroxyurea is the main disease-modifying therapy used in individuals with SCA and exerts its effects primarily by increasing fetal hemoglobin, which in turn, results in decreased production of sickled hemoglobin, a decrease in incidence of vaso-occlusive events, and an increase in total hemoglobin concentration from decreased hemolysis and prolonged RBC survival. Hydroxyurea has been shown to be effective at preventing vaso-occlusive episodes in adults and children as young as nine months of age.123–125 Further studies have shown not just a reduction in painful events but also multiorgan protection. 126 Thus, the NHLBI guidelines recommend offering hydroxyurea to all individuals with the higher risk genotypes of HbSS and HbS-0β thalassemia beginning in infancy. 36
Emerging evidence suggests that the beneficial effects of hydroxyurea extend to slowing the progression of cognitive deficits. Nottage et al. 117 observed a lower-than-expected rate of new SCI development over three to six years in children with SCD who were started on hydroxyurea. In addition, neuroimaging studies show that children with SCD on hydroxyurea had evidence of lower OEF and percentage of white matter at risk for ischemia than those not on hydroxyurea, 118 suggesting that hydroxyurea treatment might result in a reduction in cerebral metabolic stress. Patients on hydroxyurea also had normal and stable cerebral oxygen saturation values compared to the abnormal and progressive worsening seen in children with SCA not on hydroxyurea.72,127 Finally, children with SCA treated with hydroxyurea from a young age (mean age at start = 2 years) scored no differently on a brief neuropsychological battery compared to unaffected race, sex, and age-matched peers. 119 In addition, a cross-sectional analysis of the most recently completed neuropsychological batteries on children with SCA enrolled in the St. Jude longitudinal cohort study showed that an earlier age at starting hydroxyurea was associated with higher scores on measures of cognitive function, after accounting for social vulnerability and treatment duration; a primary limitation of this study was due to its cross-sectional nature, as an assessment of cognitive function over time in those on hydroxyurea versus not on therapy could not be completed. 13
Although the mechanism by which treatment with hydroxyurea results in improvement in cognitive function is not exactly known, it is hypothesized to most exert its effect through a similar manner to that proposed by chronic transfusions via an increase in cerebral arterial oxygen content. 118 Indeed, an increase in total hemoglobin concentration, but not fetal hemoglobin percentage, has been associated with lower cerebral metabolic stress and improved executive function.116,118 The increase in total hemoglobin concentration and improved oxygen saturation ensure more appropriate tissue oxygen delivery, thereby decreasing both the amount of oxygen extracted within the deep white matter and the risks for tissue ischemia and infarction.
Hematopoietic stem cell transplant and gene therapy
Hematopoietic stem cell transplant (HSCT) can be curative for individuals with SCD. Matched sibling donors offer the most favorable outcomes; in addition, even converting an affected individual from SCD to sickle cell trait (as siblings have a 50% chance of carrying a sickle trait) still results in cure. 128 However, until recently the effects of transplant on neurovascular disease were unknown. A small study of 10 children with SCD (both HbSS and HbS-β+ thalassemia with severe phenotypes) who completed MRI 1–3 months before and 12–24 months after HSCT had normalization of both elevated OEF and CBF compared to healthy sibling controls. 79 Importantly, the cohort that underwent HSCT had a greater decline in OEF and CBF compared to the decline seen after transfusion in a cohort with SCA on CTT; the HSCT cohort also had a significantly higher median hemoglobin (12.4 g/dL) versus the CTT cohort (10.6 g/dL), leading the authors to hypothesize that the greater decline in markers of cerebral metabolic stress was due to a greater relaxation of the cerebral compensatory mechanisms due to a greater restoration of oxygen delivery than that achieved with CTT. 79 However, although HSCT may offer improvements in cerebral metabolic stress, the natural history of pre-existing cerebral vasculopathy is still unknown. In a systematic review of 45 individuals with SCD with evidence of cerebrovascular abnormalities prior to HSCT and who had additional imaging after transplant with 36–72 months of follow-up, 16% had evidence of progressive neurovascular abnormalities, suggesting that HSCT cannot reverse previously existing vasculopathy. 129 When considered from a more general perspective, HSCT is not without significant risk for long-term morbidity and mortality, is often cost-prohibitive, and the availability of matched donors is limited; for these reasons, HSCT is typically recommended only in young children with a matched sibling. 130
Although a full review of the development of gene therapy in SCD is outside the scope of this article, several approaches to curing SCD through genetic modification have been pursued in recent years with the goal to either replace the abnormal beta globin gene or alternatively to induce fetal hemoglobin production. Specifically, both the use of lentivirus vectors and genome editing with clustered regularly inter-spaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) are currently undergoing phase III trials (NCT04293185 and NCT05329649 for the lentivirus and CRISPR/Cas9 trials, respectively). Although the effects on cerebrovascular disease will not be known for years, gene therapy will likely mitigate cerebral metabolic stress by a similar mechanism to that of HSCT with normalization of the hemoglobin concentration and thus relaxation of compensatory mechanisms.
Experimental agents
Currently, no therapies are under investigation to target specifically SCD-related cognitive decline. Rather, one can look theoretically to similar causes of cognitive impairment (such as vascular cognitive impairment [VCI, also known as vascular dementia] and neuroinflammation) to identify potential novel therapeutics. Although approved for Alzheimer’s disease, in randomized controlled trials, the cholinesterase inhibitors donepezil and galantamine showed a slight decrease in the rate of cognitive decline in VCI;131–134 however, the actual clinical relevance of this is less certain.
135
The N-methyl-
Another potential therapeutic target is the contributors to neuroinflammation, given the likely role the latter plays in SCD-related cognitive decline. Unfortunately, many potential therapeutic agents remain at the preclinical stage across most neurodegenerative disorders.140,141 In a preclinical SCD mouse model, the tetracycline antibiotic minocycline was shown to decrease the density of activated resident and bone marrow–derived microglia with a concurrent improvement in structural plasticity in the hippocampus. In addition, treated SCD mice had significant improvements on learning and memory tasks compared to untreated mice; the treated mice scored no differently than age and sex-matched controls. 15 The largest human trial to date, the Minocycline in Alzheimer Disease Efficacy (MADE) Trial, evaluated the role of minocycline at two dosing strategies to slow the rate of cognitive decline in individuals with mild Alzheimer’s disease over 24 months of follow-up; although no statistically significant difference was observed when comparing the patients treated at either minocycline dose versus placebo, a trend toward a significantly lower rate of decline was seen when comparing the higher-dose versus low-dose minocycline therapy. 142 In addition, the study used the standardized Mini-Mental State Examination (MMSE) as their cognitive outcome measure; however, the MMSE has very limited ability to diagnose mild cognitive impairment and does not actually examine executive function.143,144 Another potential target of neuroinflammation was based on the observation that individuals with Alzheimer’s disease and higher baseline levels of the proinflammatory cytokine tumor necrosis factor alpha (TNF-α) had a four times higher rate of cognitive decline over six months than those with low baseline TNF-α levels. 145 A phase II safety and tolerability trial of the TNF-α inhibitor etanercept in individuals with mild/moderate Alzheimer’s showed no difference in adverse events versus placebo, but also no change in cognitive measures over six months. 146 If this were to show efficacy in a larger trial, etanercept may be translatable to SCD given that individuals with SCD have significantly higher levels of TNF-α compared to unaffected individuals. 147
Neurological complications in non-SCA genotypes
Although the vast majority of data related to neurological complications and cognitive deficits are in individuals with the more severe SCA genotypes of HbSS and HbS-0β thalassemia, individuals with the non-SCA genotypes, particularly HbSC and HbS-β+ thalassemia, are also at risk for certain neurological complications and cognitive decline. Unlike in SCA, based on observations from the natural history studies, individuals with HbSC and HbS-β+ thalassemia are not at significantly higher risk for ischemic or hemorrhagic stroke with an incidence of 2% by age 20 years for individuals with HbSC. 7 Thus, the non-SCD genotypes were included in neither the trials that correlated a TAMMV TCD velocity of at least 200 cm/s with elevated stroke risk nor in the STOP trials.26–29,115 In addition, a more recent study suggested that on average, individuals with HbSC have lower CBF velocities in the Circle of Willis than individuals with the SCA genotypes; thus, a lower TCD velocity threshold may be required to predict stroke risk. 148 However, to date, this has not been investigated further. Children with the non-SCA genotypes also may develop SCI, but at a much lower prevalence than children with SCA; the latter ranges from 5.8% to 13.5% in HbSC149,150 and only 5% in HbS-β+ thalassemia (although based on a sample size of only 20 children in the CSSCD). 149 The rates of SCI progression and/or new infarct formation are unknown in this population.
Data from both the CSSCD and St. Jude longitudinal lifetime cohort study of SCD suggest that children with HbSC and HbS-β+ thalassemia also have evidence of cognitive deficits on neuropsychological testing. Wang et al. 54 found no difference in performance nor rate of change on measures of executive functioning and attention in children with HbSC compared to the SCA genotypes with anatomically normal-appearing brain MR imaging; however, the actual testing scores were not reported for these 118 children. In a cohort of 43 adolescents with HbSC and HbS-β+ thalassemia enrolled in the St. Jude longitudinal lifetime cohort study of SCD, a cross-sectional analysis of their cognitive performance revealed an average FSIQ of 86 with 19% of the cohort scoring as impaired (below the 5th percentile) on a standard neuropsychological battery. 151 However, in contrast to no difference in the rate of change in scores over time compared to the SCA genotypes in the CSSCD, using the same cohort, Heitzer et al. 13 found no association between increasing age and cognitive performance for children and adolescents with HbSC and HbS-β+ thalassemia nor an increase in the rate of cognitive impairment (i.e. scoring below the 5th percentile) with age for these genotypes compared to the SCA genotypes. Given that individuals with HbSC and HbS-β+ thalassemia typically have higher baseline hemoglobin concentrations and less hemolysis than those with the SCA genotypes, the pathophysiology of neurological complications and cognitive deficits in the non-SCA genotypes remains unclear.
Conclusions
Children and adults with SCD are especially susceptible to cognitive dysfunction, even in the absence of overt cerebral injury. The etiology of this dysfunction is multifactorial with contributions from SCD-related overt and covert cerebral injury, chronic anemia and hypoxemia, and sociodemographic factors. These cumulative and progressive changes negatively impact the lives of affected individuals via difficulties with academic achievement and later job attainment. Although chronic transfusion therapy and hydroxyurea have been shown to be helpful, both have limitations that have also limited their use. Thus, additional studies are needed to identify novel therapeutic targets and interventions that specifically focus on cognitive deficits in individuals with SCD.
Footnotes
Authors’ Contributions
KAK and JG wrote the first draft of the manuscript. HIH edited the manuscript and gave final approval.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors KK and JG received no financial support for the research, authorship, and/or publication of this article. The author HIH received support from grants from the National Institutes of Health, R01HL138423, R01HL156024, and R01AG072592.