
Abstract
Previous studies have shown that cardiomyocytes in the subendocardial region of myocardium survive from ischemic insult. This study was undertaken to explore possible mechanisms for the survival of these cardiomyocytes, focusing on changes in endothelial cells (ECs) and blood supply. C57/B6 mice were subjected to permanent ligation of left anterior descending (LAD) coronary artery to induce myocardial ischemia (MI). The hearts were harvested at 1, 4, and 7 days post MI and examined for histological changes. It was found that the survival of cardiomyocytes was associated with a preservation of ECs in the subendocardial region, as revealed by EC-specific tdTomato expression transgenic mice (Tie2tdTomato). However, the EC selective proteins, PECAM1 and VEGFR2, were significantly depressed in these ECs. Consequently, the ratio of PECAM1/tdTomato was significantly decreased, indicating a transformation from PECAM1+ ECs to PECAM1− ECs. Furthermore, EC junction protein, VE-cadherin, was not only depressed but also disassociated from PECAM1 in the same region. These changes led to an increase in EC permeability, as evidenced by increased blood infiltration in the subendocardial region. Thus, the increase in the permeability of ECs due to their transformation in the subendocardial region allows blood infiltration, creating a unique microenvironment and ensuring the survival of cardiomyocytes under ischemic conditions.
Impact statement
Cardiomyocytes in the subendocardium can survive from ischemic insults. Understanding the mechanism by which these cells escape the fatal attack is critically important for strategic development to promote myocardial regeneration. This study reveals a preservation and transformation of endothelial cells in the subendocardial region, which allows blood infiltration under ischemic conditions. This in turn creates a unique microenvironment, leading to the survival of cardiomyocytes. This self-rescuing mechanism has significant implications for myocardial regeneration.
Introduction
Coronary artery occlusion dampens cardiac microvasculature network,1,2 leading to massive loss of cardiomyocytes, except those in the subendocardial region of the myocardium.3,4 The microvascular network supplies oxygen and nutrients to the heart and preserves the viability and normal function of cardiomyocytes.5,6 We previously found that the survival of cardiomyocytes in the subendocardial region under ischemic conditions was associated with a remodeling of extracellular matrix (ECM).3,7 In particular, a significant increase in lysyl oxidase (LOX), a copper-dependent amine oxidase catalyzing the cross-linking of collagens, was involved in the ECM remodeling. The ECM remodeling made an electrical disconnection between the surviving cardiomyocytes. Inhibition of LOX disturbed the ECM remodeling and reduced the surviving cardiomyocytes under ischemic conditions. 3
Endothelial cells (ECs) are major vascular constituents and necessary for the transport of oxygen and nutrients.8,9 Coronary artery occlusion leads to profound changes in microvascular and EC functions. 10 In the clinical approach, intra-arterial thrombolysis is the most efficient way to improve the ischemic microenvironment and to rescue injured cardiomyocytes from acute myocardial ischemia (MI). 11 At present, revascularization is considered to be one of the most important methods to promote myocardial regeneration. 12 Co-transplanting micro-vessels and pluripotent stem cell–derived cardiomyocytes helps cardiomyocyte survival in the ischemic myocardium. 13 It is, therefore, worthwhile that exploring changes in the microvascular network may be relevant to cardiomyocyte survival in the subendocardial region.
The dynamic changes in ECs reflect the alteration in several functional proteins, such as platelet EC adhesion molecule 1 (PECAM1).14,15 PECAM1 is involved in the regulation of EC adhesion to form a consecutive endothelial barrier. 14 PECAM1 deficiency impairs the integrity of EC junctions, facilitating EC transformation and increasing vascular permeability.14,16 In addition, the type 2 receptor for vascular endothelial growth factor (VEGFR2) is involved in EC proliferation and migration. VEGFR2 mediates VEGF signaling cascades, activating DNA duplication and cell division. 17 In response to ischemic insult, these functional proteins in ECs are altered, resulting in EC activation as well as transformation, and myocardial microvascular remodeling.
This study was undertaken to explore changes in ECs in response to ischemic insult and their relation to cardiomyocyte survival in the subendocardial region. We found that ECs underwent a transformation, leading to an increase in their permeability, and allowing blood infiltration to constitute a unique microenvironment. This likely contributes to the survival of cardiomyocytes in the subendocardial region.
Materials and methods
Animal and animal care
Male C57/B6 mice (8–10 weeks old, weighing 18–22 g) were obtained from Chengdu Da-Shuo Experimental Animal Breeding and Research Center (Sichuan, China). Tie2tdTomato mice were achieved by crossing Tie2-Cre mice (Jackson’s laboratory, JAX No. 008863) with Rosa26-tdTomato mice carrying the loxP-flanked (“floxed”) stop cassette-controlled fluorescent marker tdTomato gene. Mice were fed standard chow (5C02, LabDiet, USA) and tap water ad libitum. Mice were acclimated to the experimental conditions for at least 1 week before surgery. All animal procedures were approved by the Institution Animal Care and Use Committee (IACUC) at Sichuan University, West China Hospital, following the guidelines of the US National Institutes of Health.
Genotypic identification
The tails of the transgenic mice were excised at the age of 6 to 7 weeks for DNA extraction. Genotyping was then performed by a standard PCR method using Cre-positive primers (5′-CACCCTGTTACGTATAGCCG-3′; 5′-GAGTCATCC TTAGCGCCGTA-3′), Cre-negative primers (5′-CCTAGGCACCAGGGTGTGAT-3′; 5′-TCACGGTTGGCCTTAGGGTT-3′), and tdTomato primers (pr1590: 5′-AAGGGAGCTGGCAGTGGAGTA-3′; pr1591: 5′-CCGAAAATCTGTGGGAAGTC-3′; pr1592: 5′-GGCATTAAAGCAGCGTATCC-3′; pr1593: 5′- CTGTTCCTGTACGGCATGG-3′).
Mouse model of MI
C57/B6 mice were randomly divided into sham-operated (n = 45) and MI (n = 66) groups. After genotyping the Tie2tdTomato mice, the positive mice were also randomly divided into sham (n = 3) and MI (n = 6) groups. The MI group was subjected to left anterior descending (LAD) artery ligation as described previously. 3 Briefly, mice underwent open chest surgery after anesthesia by isoflurane inhalation. The LAD was exposed, and a 7-0 suture was pierced beneath. Then, the LAD was ligated to induce MI, and the mice revived shortly after sternal closure. The sham group underwent the same procedure as the MI group, except for LAD ligation. Of these mice, 112 survived until the time of sacrifice, including 105 C57/B6 mice and 7 Tie2tdTomato mice, and these mice were used for further investigations.
Tissue preparation
Mice were deeply anesthetized with isoflurane (5%) inhalation and euthanized in a CO2-rich cage. Heart samples were collected for histological analysis. Briefly, the chests were opened, and the hearts were removed immediately. After washing in precooled saline (4℃), the atria were removed. For histological analysis, the hearts of mice were dissected along the ligature, embedded in optimal cutting temperature compound gel (Leica, Germany), and frozen in liquid nitrogen for serial frozen sections. The tissues were sectioned at 4 μm intervals and stored at –20℃ for immunofluorescence staining. The remaining hearts were fixed in 4% paraformaldehyde, dehydrated in gradient alcohol, embedded in paraffin, and then cut into 2.5 μm sections for hematoxylin and eosin (HE) staining.
Hypoxyprobe detection
The hypoxyprobe-1 Omni kit (Hypoxyprobe, USA) was used to detect the oxygen content in the myocardium after ischemic injury according to the manufacturer’s protocol. Hypoxyprobe-1 is widely used to detect hypoxic conditions in animal models. Once injected into an animal, the probe is rapidly distributed to all tissues in the body, while it only forms an adduct with proteins in cells that have O2 concentrations < 14 μM (equivalent to a partial oxygen pressure of 10 mmHg at 37°C). One hour before sacrifice, the mice were intraperitoneally injected with a 10 mg/mL solution (60 mg/kg) of hypoxyprobe-1 (pimonidazole HCl). After that, the mice were sacrificed by cervical dislocation to abruptly cut off the blood circulation. Then the chests were opened, and the hearts were removed immediately for dissection. The hearts were sectioned at 4μm intervals and labeled with rabbit anti-hypoxyprobe antibody (PAb2627AP), which is included in the kit, followed by labeling with an Alexa-Fluor-conjugated secondary antibody. Co-injection of hypoxyprobe with lectin was performed as follows: 1 hour before sacrifice, the mice were intraperitoneally injected with hypoxyprobe-1; and 5 min before sacrifice, the mice were tail intravenously injected with lectin. Hearts collected and stained as above.
Histological and immunofluorescence staining
Tissue sections were fixed in precooled 4% paraformaldehyde for 15 min and then washed with phosphate-buffered saline (PBS) for 3 min, three times. The sections were blocked with 2% bovine serum albumin (BSA) for 1 h at 37℃. Primary antibodies were incubated at 37°C for 1 h, followed by incubation at 4°C overnight. The primary antibodies used were as follows: TNNI3 (ab56357, Abcam, 1:200), RFP (600-401-379, Rockland, 1:1000), PECAM1 (AF3628, R&D, 1:100), VEGFR2 (9698, Cell Signaling Technology, 1:100), VE-cadherin (ab33168, Abcam, 1:200), and HIF-1α (AF1935, R&D, 1:100). After incubation, the sections were washed with PBS for 10 min, three times. Then, the secondary antibodies were incubated at 37℃ for 1 h at a concentration of 1/1000. The secondary antibodies used were as follows: Alexa Fluor 488 goat anti-mouse (Thermo Fisher, A-11001), Alexa Fluor 488 donkey anti-goat (Thermo Fisher, A-11055), Alexa Fluor 488 goat anti-rabbit (Thermo Fisher, A-11008), Alexa Fluor 568 goat anti-rabbit (Thermo Fisher, A-11011), and Alexa Fluor 647 chicken anti-rabbit (Thermo Fisher, A-21443). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI, Sigma, D9542). The blank control was incubated with 2% BSA instead of primary antibodies. All sections were examined by a laser confocal microscopy (ECLIPSE Ti A1, Nikon). All sections were taken approximately 100 μm below the ligature, and at least three fields for each section were captured for each animal unless specifically mentioned.
Apoptosis detection
EC apoptosis was detected in situ using a terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end-labeling assay (TUNEL; 11767305001, Roche, USA) following the manufacturer’s instructions. Briefly, the tissue sections were fixed with 4% paraformaldehyde for 15 min, washed with PBS for 3 min, two times and treated with a precooled permeabilization solution (0.1% Triton X-100) for 2 min. After washing with PBS for 3 min, two times, the labeling reaction was carried out in a solution containing terminal deoxynucleotidyl transferase and fluorescein-dUTP at 37°C for 1 h. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI, Sigma, D9542). TUNEL staining was examined by a laser confocal microscope (ECLIPSE Ti A1, Nikon).
Measurement of blood perfusion
Fifty micrograms of fluorescein isothiocyanate (FITC)-labeled Lycopersicon esculentum (tomato) lectin (MP6311, MKbio) diluted in 100 μL of sterilized PBS was injected into the mice through the tail vein 5 min before sacrifice according to a previous study. 2 Heart tissues were embedded in optimal cutting temperature compound gel (Leica, Germany), frozen in liquid nitrogen, and cut into 5 or 100 μm sections for visualization of blood infusion and three-dimensional (3D) construction using a laser confocal microscope (ECLIPSE Ti A1, Nikon). The ischemic area (IA) was recognized by a reduction in lectin infusion compared with the sham-operated control. The percentage of lectin-positive volume relative to the total tissue volume indicates the extent of blood perfusion for each sample, and was calculated using ImageJ.
Statistical analysis
ImageJ was applied to analyze the stained images. All data are presented as the mean ± SD. GraphPad Prism 7.0 was applied to perform statistical analysis. Two-way analysis of variance (ANOVA) was used to evaluate different times after LAD ligation and sham operation, and Student’s t-test was used to compare the difference between the MI group and the sham-operated group. The value p < 0.05 was considered statistically significant.
Results
Hypoxic location in the subendocardial region of ischemic myocardium
Overall, there was a significant loss of cardiomyocytes, as detected by HE staining (see Figure 1(a)) and cardiomyocyte-specific marker TNNI3 (Cardiac Troponin I Type 3) staining (see Figure 1(b)) after MI. However, cardiomyocytes in the subendocardial region of the ischemic myocardium survived from day 1 to day 7 post ischemia (see Figure 1). The use of a hypoxyprobe determined that the hypoxic transition took place right after ischemia, sustained on day 1 post ischemia, and declined thereafter (see Figure 2). Importantly, in the subendocardial region, the hypoxyprobe binding was mostly located at the boundary of areas occupied by surviving cardiomyocytes (see Figure 2). Correspondingly, staining for the key hypoxia-responsive transcription factor (hypoxia-inducible factor-1 alpha, HIF-1α) showed that HIF-1α was less detectable in the subendocardial region compared to the other regions of the IA (see Supplemental Figure 1).
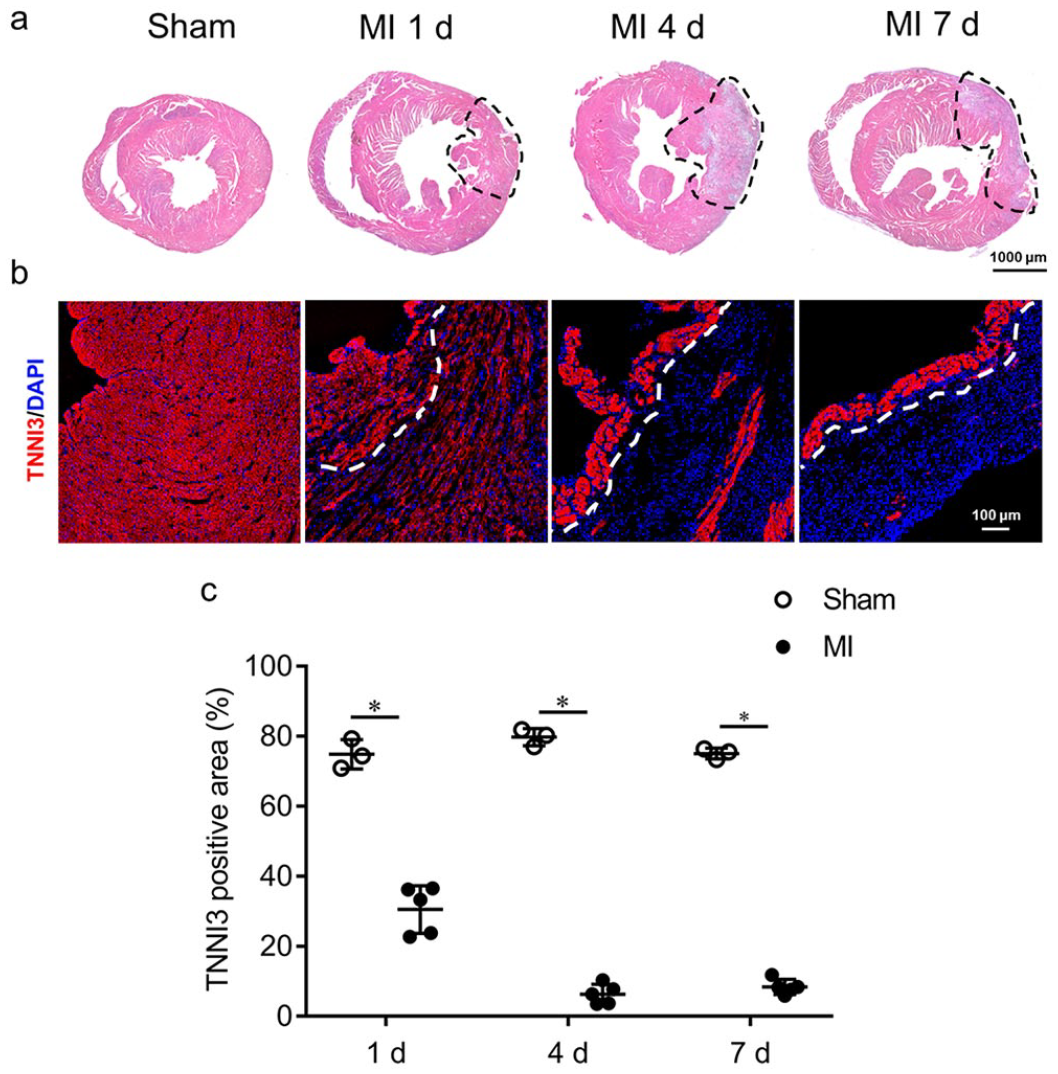
Survivalof cardiomyocytes in the subendocardium of ischemic myocardium.
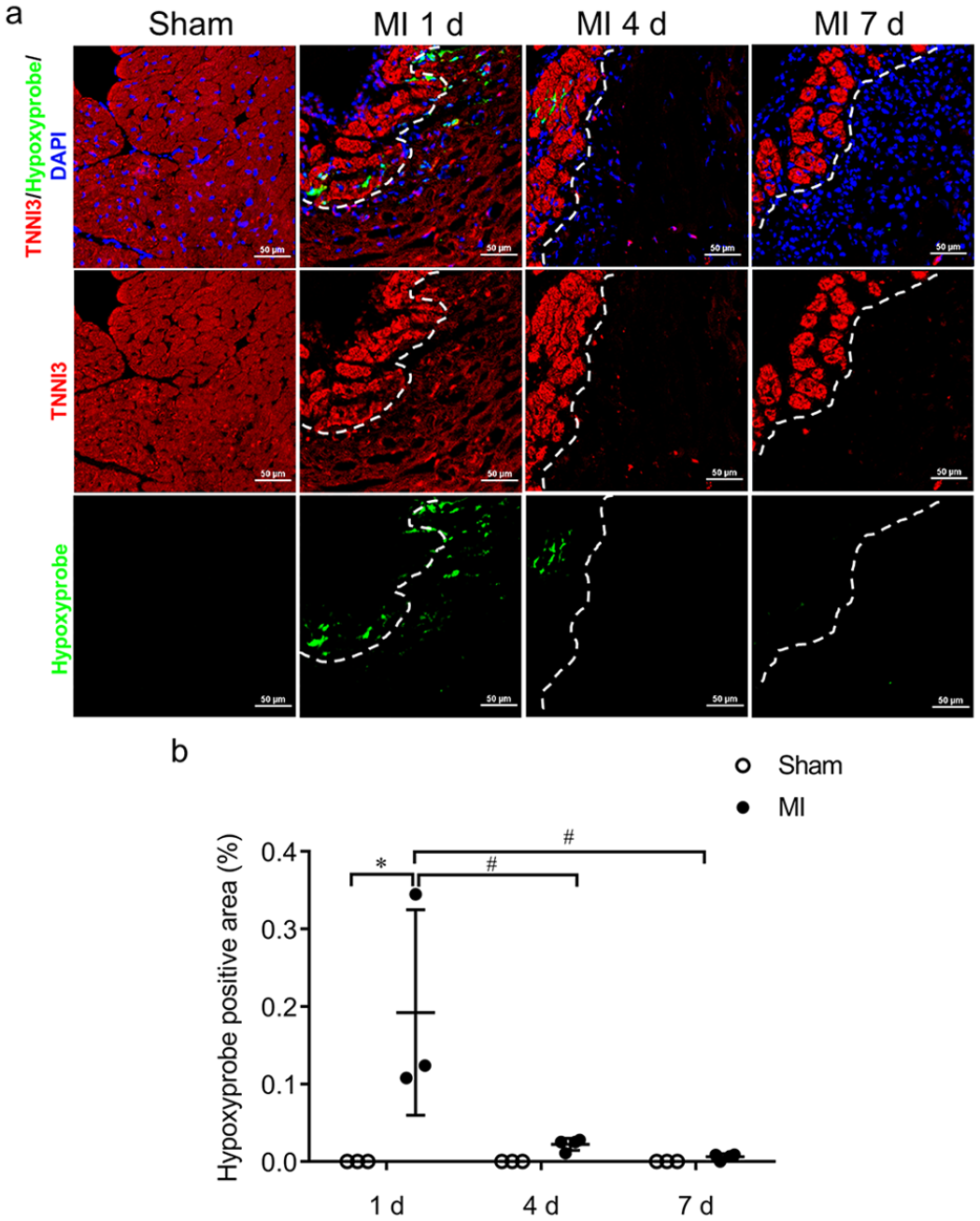
Detection of hypoxyprobe in the subendocardium of ischemic myocardium.
Transformation of ECs in the subendocardium after ischemic insults
We determined the changes of a selective protein of ECs, PECAM1 (see Figure 3). Reduced PECAM1+ ECs (see Figure 3(a) and (b)) and downregulation of PECAM1 expression (see Figure 3(a) and (c)) were observed in the ischemic myocardium on day 1 post MI,whereas PECAM1 expression was recovered on day 4 and 7 post MI (see Figure 3(c)). VEGFR2, another EC marker, was also dramatically decreased on day 1 post MI (see Supplemental Figure 2). In contrast to the depressed EC selective marker proteins, TUNEL staining indicated that the ECs in the subendocardial region did not undergo apoptosis (see Figure 3(d) to (f)).
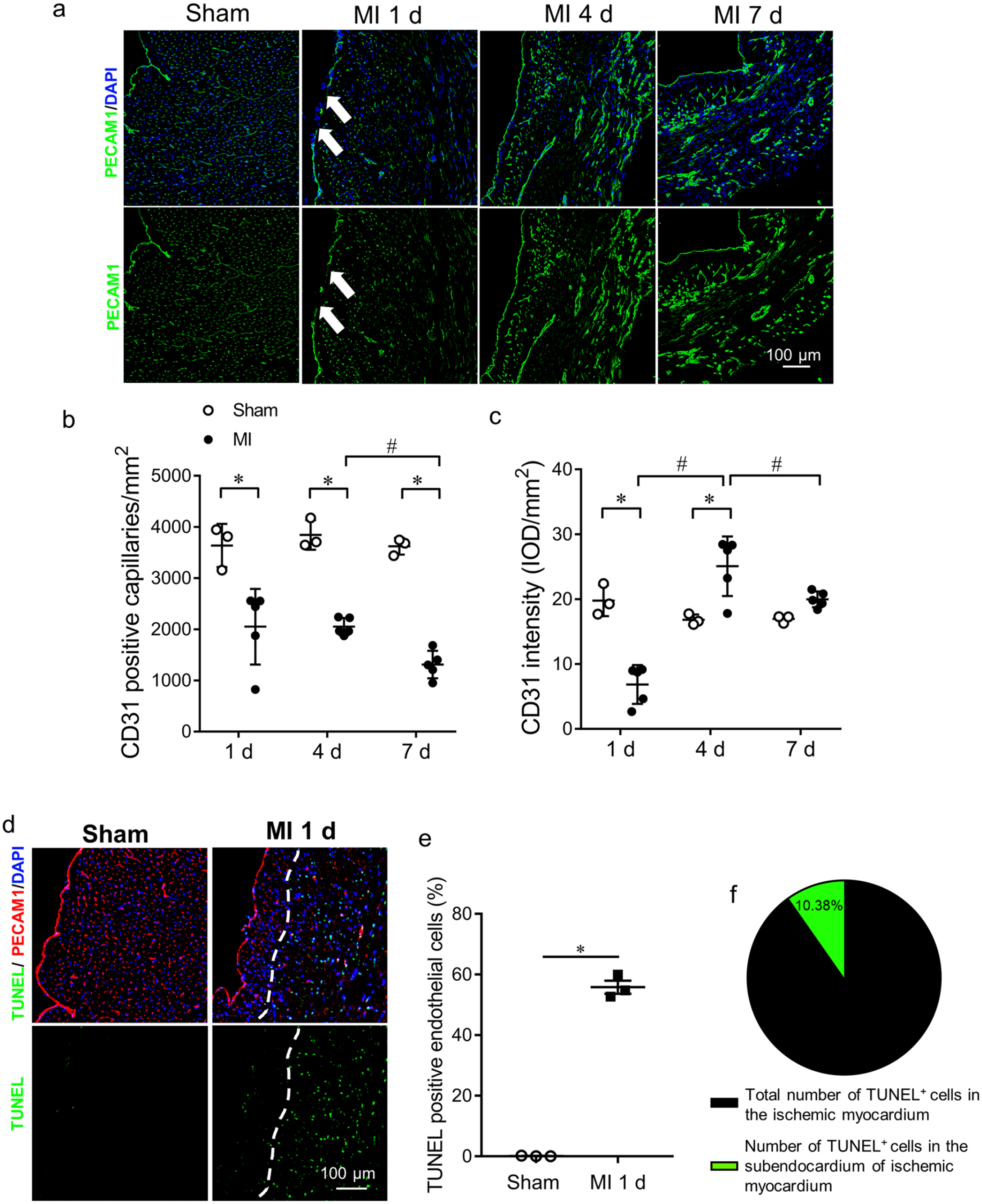
Decrease in endothelial cells after ischemic insults.
To investigate whether the suppression of EC marker proteins results from a reduction of the number of ECs or from the reduced expression in the same ECs, we used Tie2tdTomato transgenic mice, in which ECs were specifically labeled by red fluorescent protein (see Figure 4(a), Supplemental Figures 3 and 4). It was observed that tdTomato-positive ECs (tdTomato+ ECs) decreased in most regions of the ischemic myocardium but remained the same in the subendocardial region on day 1 post-MI (see Figure 4(b) and (c)). We then calculated the ratio of PECAM1/tdTomato to reflect the change of the surviving ECs. This ratio was significantly decreased, indicating a transformation from PECAM1+ ECs to PECAM1- ECs in the subendocardial region (see Figure 4(d) and (e)). We also examined the EC junction protein, VE-cadherin, which was also decreased. Furthermore, co-immunostaining of VE-cadherin and PECAM1 revealed that these two proteins became disassociated in the ischemic myocardium, in contrast to being co-localized in the sham-operated controls (see Figure 4(f) and (g)).
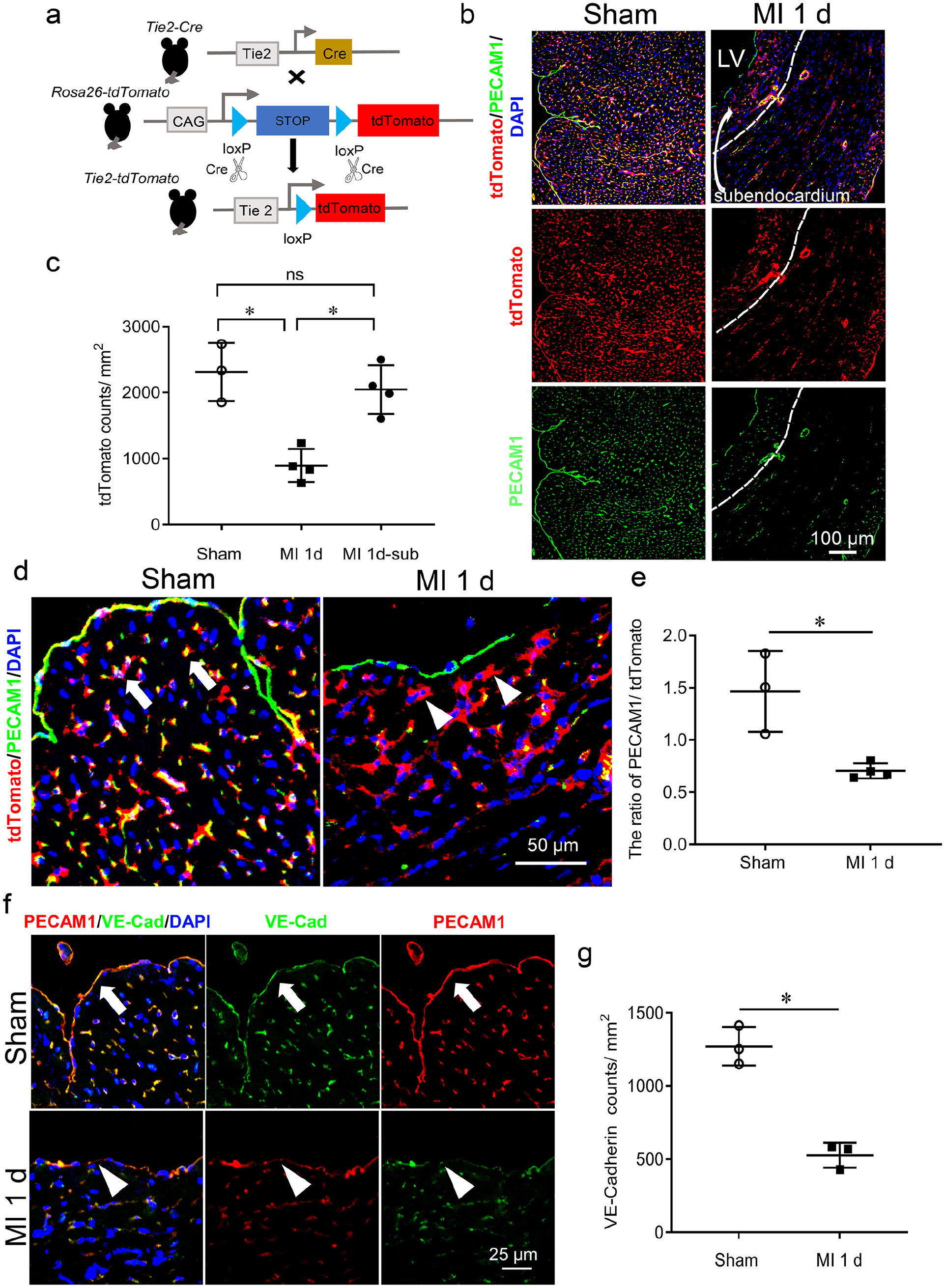
Reservation of endothelial cells and changes in the endocardial junction in the subendocardium of ischemic myocardium.
Blood infiltration in the subendocardial region of ischemic myocardium
With regard to the changes of the endothelial junction, we detected blood infiltration in the subendocardial region of the ischemic myocardium. Intravenous injection of FITC-lectin was used to reflect blood perfusion. LAD coronary artery ligation blocked FITC-lectin perfusion to most IAs on day 1 post MI, with the exception of the subendocardial region of the ischemic myocardium (see Figure 5(a) to (c)), indicating an increase in the permeability of the vasculature in the subendocardial region. Co-injection of hypoxyprobe with FITC-lectin revealed that the blood-infiltrated subendocardial region was much less hypoxic on day 1 post MI, as evidenced by the separation of the lectin perfusion area and the hypoxyprobe binding area (see Figure 5(d)).
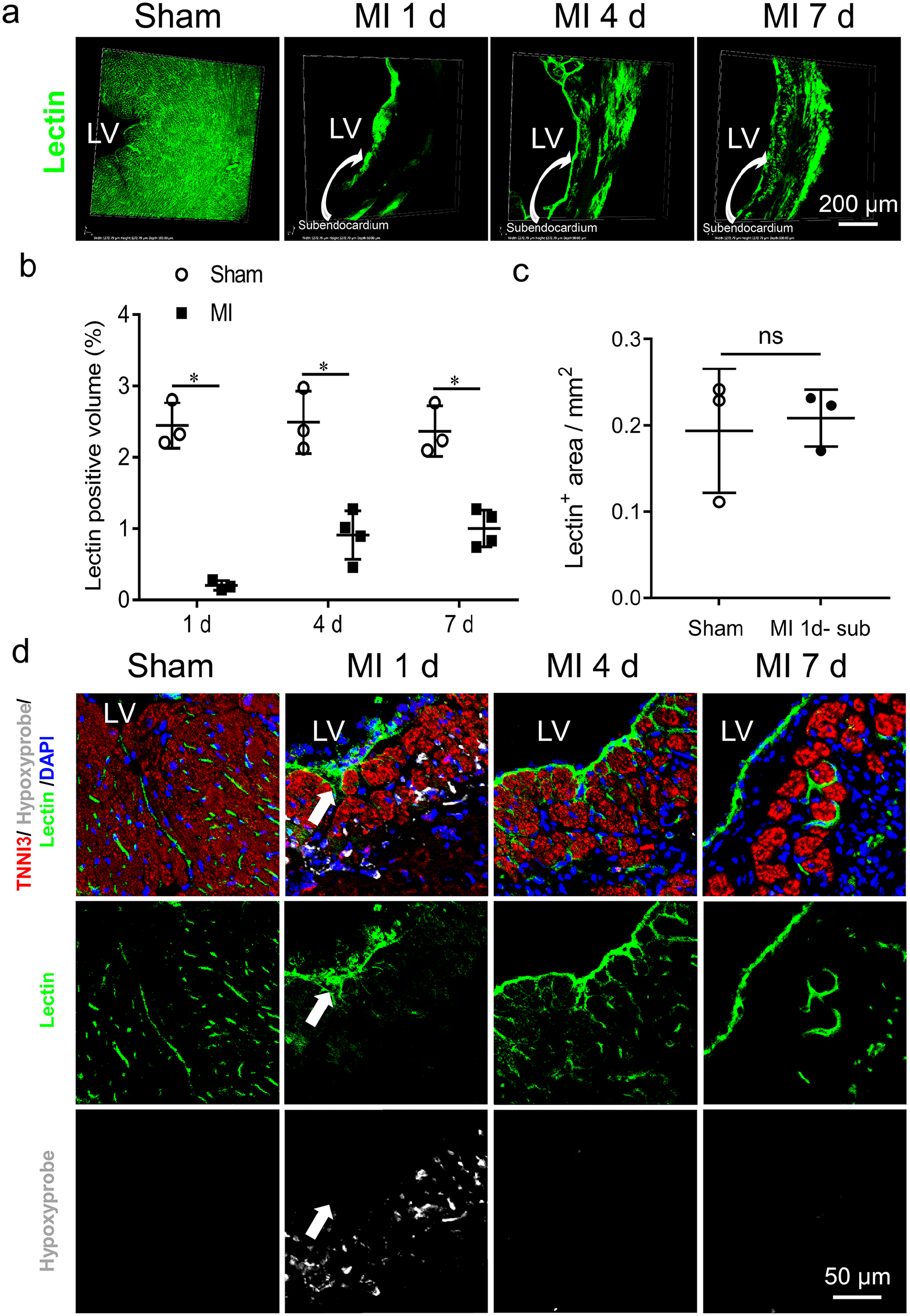
Changes in blood supply in the subendocardial region of ischemic myocardium.
Discussion
Cardiomyocyte survival in the subendocardial region of ischemic myocardium has been observed in human18,19 and animal models.3,4,7,20 Previous studies have shown that preservation of the functional microvascular bed is vital for the long-term survival of cardiomyocytes. 4 However, the reasons these cardiomyocytes may escape ischemic injury have not been elucidated. This study used transgenic mice with EC-specific labeling and investigated the role of endothelial alterations in cardiomyocyte survival. The study identified a reservation of ECs in the subendocardial region during the early stage of MI, which underwent a transformation with an increase in their permeability. This allowed blood infiltration and created a unique microenvironment, ensuring the survival of cardiomyocytes in the subendocardium under ischemic conditions.
ECs are major constituents of blood vessels, play a vital role in the physiological function of blood vessels, and require a precise coordination of multiple functional proteins.21–23 In response to ischemic injury, ECs display various gene expression signatures, as currently revealed by single cell RNA sequencing. 24 These changes are associated with EC activation and the release of proteases, which in turn degrade the ECM and contribute to the ECM remodeling. 25 We found that during the ECM remodeling, LOX plays a critical role in collagens deposition and reorganization. 3 On the contrary, ECM remodeling can alter the mechanical properties of the tissue and lead to changes in EC shape and migration, contributing to the formation of new blood vessels and the recovery of injured myocardium.26,27 In addition, ECM remodeling can affect the expression of EC surface molecules, which can alter EC adhesion.28,29 Understanding the dynamic changes of ECs and ECM remodeling in response to ischemic injury is important for the development of new therapeutic strategies for the treatment of heart diseases.30,31
Here, we observed changes in critical proteins in ECs, leading to an increase in vasculature permeability in the subendocardial region of the ischemic myocardium. ECs cover the cardiac chamber, which regulate vascular barrier function and control the passage of plasma proteins and circulating cells across the endothelium.9,32 Dysregulation of endothelial permeability is associated with many diseases, including inflammation and edema. 33 Thus, endothelial permeability is tightly controlled by numerous extracellular components and mediators to maintain tissue homeostasis.34,35 PECAM1, also known as CD31, belongs to the immunoglobulin family and is a critical component of the ECM for ECs, regulating the cell–cell interactions. 16 Studies have also revealed many other functions of PECAM1, including acting as a receptor in response to shear stress and mediating neutrophil adhesion.36,37
In this study, we observed that PECAM1 was significantly decreased after ischemic insult, but ECs remained viable in the subendocardial region of the ischemic myocardium. Previous studies found that the loss of PECAM1 disrupts the blood–brain barrier (BBB), allowing T-cell trafficking to the central nervous system in multiple sclerosis. 38 We found that PECAM1 partially disappeared, thus leaving an endothelial gap and a disruption of the EC junction in the endocardium. VE-cadherin is another important component of EC junctions,34,39 and its concomitant depression with the reduction in PECAM1 in the surviving ECs further indicates the disruption of EC junctions in the subendocardial region. As the pathological process progresses, angiogenesis occurs in the ischemic myocardium, which can partially compensate for the reduced blood supply.28,31 The restoration of blood supply might help to restore the expression of PECAM1 in ECs, leading to the recovery of endothelial permeability. Thus, the dynamic change in endothelial permeability and blood infiltration in the subendocardial region nourishes cardiomyocytes and ensures their survival in the early stage of MI.
Apoptotic cells were rarely detected in the subendocardial region, and this might be related to the unique microenvironment of the subendocardial region. 3 Although PECAM1 depression can cause EC apoptosis, 40 we did not observe an association between PECAM1 depression and EC apoptosis on day 1 after ischemic insult in the subendocardial region. Thus, increased permeability of ECs due to the disruption of the cell–cell conjunction would be responsible for blood infiltration for the survival of cardiomyocytes in this region. VEGFR2 is a receptor corresponding to VEGF that activates cell proliferation and growth. 17 We found that VEGFR2 was dramatically depressed in the surviving ECs, suggesting a limited EC proliferation in the early stage of ischemia.
Changes in surviving ECs and thereafter an increase in the permeability of the vasculature would therefore constitute a mechanism for the survival of cardiomyocytes in the subendocardial region. On the contrary, cardiomyocytes not only survive in the microenvironment supported by ECs, but also in turn promote angiogenesis through paracrine effects under the ischemic condition. 41 Thus, cardioprotective cross-talk between cardiomyocytes and ECs ensures the continuous progression of myocardial regeneration. 42 This endogenous self-rescuing mechanism for cardiomyocytes in the subendocardial region should be further investigated for a better understanding of myocardial regeneration.
In summary, we demonstrate that there are surviving but transformed ECs in the subendocardial region in response to ischemic insult, leading to an increased permeability of the vasculature to allow blood infiltration. This blood infiltration improves the microenvironment, likely leading to the survival of cardiomyocytes in the subendocardial region of the ischemic myocardium. This self-rescuing mechanism for myocardial survival from ischemic injury can therefore be considered great importance for an understanding of myocardial regeneration.
Supplemental Material
sj-pdf-1-ebm-10.1177_15353702231194344 – Supplemental material for Alteration of endothelial permeability ensures cardiomyocyte survival from ischemic insult in the subendocardium of the heart
Supplemental material, sj-pdf-1-ebm-10.1177_15353702231194344 for Alteration of endothelial permeability ensures cardiomyocyte survival from ischemic insult in the subendocardium of the heart by Qing Chu, Xin Song, Ying Xiao and Y James Kang in Experimental Biology and Medicine
Footnotes
Authors’ Contributions
YJK conceptualized the idea of this study, and all authors participated in the experimental design, interpretation of the results, and review of the article; QC, YX, and XS were involved in the experimentation; QC performed data analysis; YJK, QC, and XS wrote the article, and YJK edited and approved the final version of the article.
Declaration Of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (NSFC 8123004) and Sichuan University West China Hospital.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.