
Abstract
As an active substance isolated from the root of Morinda officinalis How., rubiadin-1-methyl ether (RBM), can improve osteoporosis due to its inhibition on osteoclastogenesis. Autophagy plays a key role in osteoclastogenesis. Our research aims to explore the relationship between RBM, autophagy, and osteoclastogenesis. Our results showed that RBM not only inhibited the differentiation level of osteoclasts and the proliferation ability of osteoclast precursors (OCPs), but also repressed the autophagic activity in OCPs (LC3 transformation and the number of autophagosomes observed by transmission electron microscopy). However, RBM-inhibited osteoclast differentiation and OCP autophagy (LC3 transformation and LC3-puncta formation) could be reversed by the application of TAT-Beclin1. Moreover, RBM administration reduced RANKL-induced p65 phosphorylation and p65 nuclear translocation in OCPs. In addition, the addition of RBM inhibited Beclin1 protein level and BECN1 (the gene form of Beclin1) mRNA level in OCPs increased by RANKL. Importantly, the reduction in the expression of BECN1 and Beclin1, LC3 transformation, and osteoclastic differentiation in OCPs caused by RBM were reversed by p65 overexpression. In conclusion, RBM may reduce the transcription of BECN1 by inhibiting the activation of nuclear factor kappa B (NF-κB) p65, thereby inhibiting Beclin1-dependent autophagy and RANKL-induced osteoclastogenesis.
Impact Statement
As a bioactive substance of Morinda officinalis How., rubiadin-1-methyl ether (RBM), can be used to treat osteoporosis due to its inhibitory effect on osteoclastogenesis. Autophagy is of great significance in osteoclastogenesis. Therefore, this study explored the relationship between RBM-regulated osteoclastogenesis and autophagic responses. Our study elucidated the inhibitory effect of RBM on osteoclast precursor (OCP) autophagy and the significance of Beclin1-dependent autophagy for RBM-inhibited osteoclastogenesis. Importantly, our experimental data described a biological process: RBM suppresses RANKL-induced transcription of autophagy gene BECN1 by inhibiting the activation of nuclear factor kappa B (NF-κB) p65, thereby suppressing Beclin1 expression and Beclin1-dependent autophagy activation, and inhibiting subsequent osteoclastogenesis. Our study not only describes for the first time the molecular mechanism of RBM-inhibited osteoclastogenesis from the perspective of autophagy, but also provides more potential clues for the treatment of RBM in osteoclastic bone loss.
Introduction
Bone integrity depends on the dynamic equilibrium between osteoblast-performed bone formation and osteoclast-exerted bone absorption. Excessive bone absorption caused by enhanced osteoclast formation leads to bone loss, thereby resulting in osteoporosis.1,2 Therefore, osteoclast is the cytological basis of osteoporosis and the main therapeutic target. The traditional Chinese herbal medicine of Rubiaceae, M. officinalis How., and its root are used in the treatment of impotence, depression, inflammatory diseases, and osteoporosis. 3 Of note, M. officinalis How. and various derivatives obtained have significant effects on the treatment of osteoporosis.4–6 Moreover, M. officinalis How. and related extracts can significantly inhibit osteoclast formation, which is the biological basis for their anti-osteoporosis function.7–10 As a natural anthraquinone compounds derived from M. officinalis How., rubiadin-1-methyl ether (RBM) also has an obvious effect on the treatment of osteoporosis, which is attributed to its anti-osteoclastogenic efficacy.7,8 However, the internal mechanism of RBM-inhibited osteoclastogenesis is still unclear, which hinders the further development of M. officinalis How. in treating osteoporosis.
As an important intracellular protection mechanism, autophagy provides energy for cell renewal by degrading various macromolecules, pathogens, and aging organelles, thus maintaining cell survival and development. Autophagy also plays an important role in the proliferation, differentiation, and death resistance of osteoclasts as well as bone resorption activity.11–14 As the core inducer of osteoclastic differentiation, receptor activator of nuclear factor kappa-B (RANKL) can stimulate autophagy in osteoclast precursors (OCPs), which is beneficial to the formation of osteoclasts.11,15 As an important autophagy regulator, Beclin1 plays a key role in autophagy activation.16,17 Furthermore, Beclin1 is also a key molecule responsible for osteoclast formation.11,15,18,19 Remarkably, RANKL can significantly promote the expression level of Beclin1. Previous study has clarified that RANKL can promote the transcription of BECN1 (the gene form of Beclin1) via the role of transcription factor NF-κB, thus inducing OCP autophagy. 15 Nevertheless, RBM can inhibit RANKL-induced p65 phosphorylation and p65 nuclear translocation in OCPs, 7 and p65 is an important component of NF-κB and controls the transcription characteristics of NF-κB. We propose a hypothesis that RBM can repress the transcription of BECN1 by inhibiting the nuclear translocation of NF-κB p65, thereby inhibiting Beclin1-related autophagy and affecting osteoclast formation. Our study revealed for the first time the internal mechanism underlying the anti-osteoclastogenic efficacy of RBM derived from M. officinalis How. from the perspective of autophagy.
Materials and methods
Extraction and induction of OCPs
The tibiae from 4- to 6-week-old C57BL/6J mice (GemPharmatech, Nanjing, China) were flushed with alfa- Minimum Essential Medium (α-MEM. Bone marrow cells were incubated in α-MEM along with 10% fetal bovine serum (FBS) + 100 U/mL penicillin + 100 μg/mL streptomycin for 24 h. Non-adherent cells were collected and incubated on dishes containing macrophage colony-stimulating factor (M-CSF) (30 ng/mL) for 3 days. The adherent cells were harvested and regarded as bone marrow–derived macrophages (BMMs), known as OCPs. 20 OCPs were cultured in a humidified environment of 37°C and 5% CO2, and then induced into mature osteoclasts.
Osteoclastic differentiation assays
OCPs (8000 cells/well) were cultured in 96-well plates in α-MEM along with M-CSF (30 ng/mL in all assays) + RANKL (100 ng/mL in all assays) for 4 days to induce mature osteoclasts. Osteoclast formation was determined by tartrate resistant acid phosphatase (TRAP) staining using a relevant kit (Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer’s protocols. TRAP-positive multinucleate cells (over three nuclei) were termed as mature osteoclasts. TRAP-positive cells over five nuclei were termed as large osteoclasts. Furthermore, the intervention concentrations of RBM were determined according to previous studies. 7 On the one hand, OCPs were treated with different concentrations of RBM (0, 0.1, 1, and 10 mM) for corresponding times determined according to specific experiments. In addition, OCPs were treated with 10 mM of RBM along with or without corresponding reagents or lentivirus with target gene to observe other experiments.
Cell proliferation assays
Cell proliferation was evaluated via cell counting kit-8 (CCK-8; Dojindo, Shanghai, China). Cells were cultured in 96-well plates at a density of 5000 cells/well, and then received different treatments. Next, CCK-8 reagents were added into each well, and cells were incubated in the darkness condition for 1 h. Eventually, the optical density at 450 nm (OD450) was measured using Varioskan Flash reader (Thermo Fisher Scientific, Waltham, MA, USA).
Lentiviral transduction of complementary DNA
Lentiviruses encoding p65 complementary DNA (cDNA) and the corresponding control vector were constructed by homologous recombination between an expression vector (EX-Puro-Lv105) and cDNA in 293 cells using construction kits (GeneCopoeia, Rockville, MD, USA) according to the manufacturer’s protocols. After 2 days, lentiviral supernatants were collected. At a multiplicity of infection (MOI) at 10, OCPs were incubated in the lentiviral fluid containing 8 μg/mL polybrene for 2 days. The infected cells were selected by puromycin (5 μg/mL). The transduction efficiency was identified via Western blotting.
Western blotting assays
The whole lysates from treated OCPs was prepared, packaged into 10% sodium dodecyl sulfate polyacrylamide gel (SDS-PAGE) and transferred to polyvinylidene fluoride membrane (PVDF). Then, the PVDFs were incubated with antibodies targeting rabbit LC3B (ab192890, 1:2000), Beclin1 (ab207612, 1:1000), phosphorylated (P-)p65 (ab76302, 1:1000), p65 (ab32536, 1:2000), Lamin B1 (ab133741, 1:10,000), and GAPDH (1:5000) (Abcam, Cambridge, UK). Horseradish peroxidase (HRP)-linked secondary antibody was used as the secondary antibody. The signals were observed using artificial exposure.
Nuclear and cytoplasm separation
The cytoplasmic and nuclear protein was extracted via nuclear and cytoplasmic protein extraction kit (Beyotime, Shanghai, China). After rinse with phosphate-buffered saline (PBS), the treated OCPs were harvested by pipetting and centrifugation. Two hundred microliters of cytoplasmic protein extraction reagent was added into the harvested cells, and cells were violently vibrated followed by ice bath for 10 min. Following high-speed centrifugation for 5 min, the supernatant was harvested to obtain the cytoplasmic protein. The residual supernatant in the remained precipitation was completely removed and 50 µL of nuclear protein extraction reagent was added into the precipitate. The nuclear suspension was violently vibrated followed by ice bath for 30 min. Subsequently, the nuclear suspension was centrifuged, and the supernatant was harvested to obtain nuclear protein. Eventually, the protein was mixed with 1× loading buffer, boiled for 10 min, and then used for Western blotting assays.
Quantitative real-time PCR assays
The total RNA from treated cells was extracted and purified by Trizol methods. cDNA synthesis and quantitative real-time polymerase chain reaction (qRT-PCR) assays were carried out according to the manufacturer’s protocols (Takara, Tokyo, Japan). The predesigned primer sequences are shown in Table 1.
Specific primer sequences for qRT-PCR.

qRT-PCR: quantitative real-time polymerase chain reaction.
In the qRT-PCR detection, the melting curve was analyzed to ensure that there was no amplification artifact. qRT-PCR was carried out using SYBR Premix Ex TaqTM kit (Takara, Tokyo, Japan) and ABI7500 PCR system (Applied Biosystems, Thermo Fisher Scientific, MA, USA).
Transmission electron microscopy analyses
The preparation, staining, and transmission electron microscopy (TEM) analyses of cell sections were carried out according to the manufacturer’s protocols. Eventually, the stained sections were observed under Hitachi 7700 transmission electron microscope (Tokyo, Japan).
Immunofluorescence assays
For the immunofluorescence staining related to LC3-puncta formation, the perforated cells were incubated with anti-rabbit LC3B antibody (ab192890, 1:200) at 4°C overnight, and then stained with fluorochrome-labeled secondary antibody for 1 h. Eventually, cell nuclei were counterstained with DAPI for 15 min, and then the images were acquired via fluorescent microscope (Olympus IX81, Tokyo, Japan).
Statistical analyses
The experiments were replicated at least three times. All data are expressed as mean ± SEM from three independent experiments. The statistical analyses were performed via one-way and two-way analysis of variance (ANOVA) using SPSS 22.0 software. Tukey test was applied for post hoc multiple comparisons of one-way and two-way ANOVA. The threshold value for P value is set to 0.05.
Results
Treatment of RBM-inhibited osteoclast differentiation and OCP proliferation
We first confirmed the effect of RBM on the proliferation ability of OCPs and osteoclast differentiation. It was observed that RBM reduced the proliferation level of OCPs in a concentration-dependent manner in the presence of osteoclastic inducer (RANKL + M-CSF) (Figure 1(A)). Moreover, RBM also inhibited the number and size of mature osteoclasts and the expression levels of osteoclast-related genes (CTSK, MMP9, and TRAP) in a concentration-dependent manner (Figure 1(B) to (G)). These results suggested that RBM has an inhibitory effect on osteoclast formation, which indicated that our pharmacological experimental system is reliable.
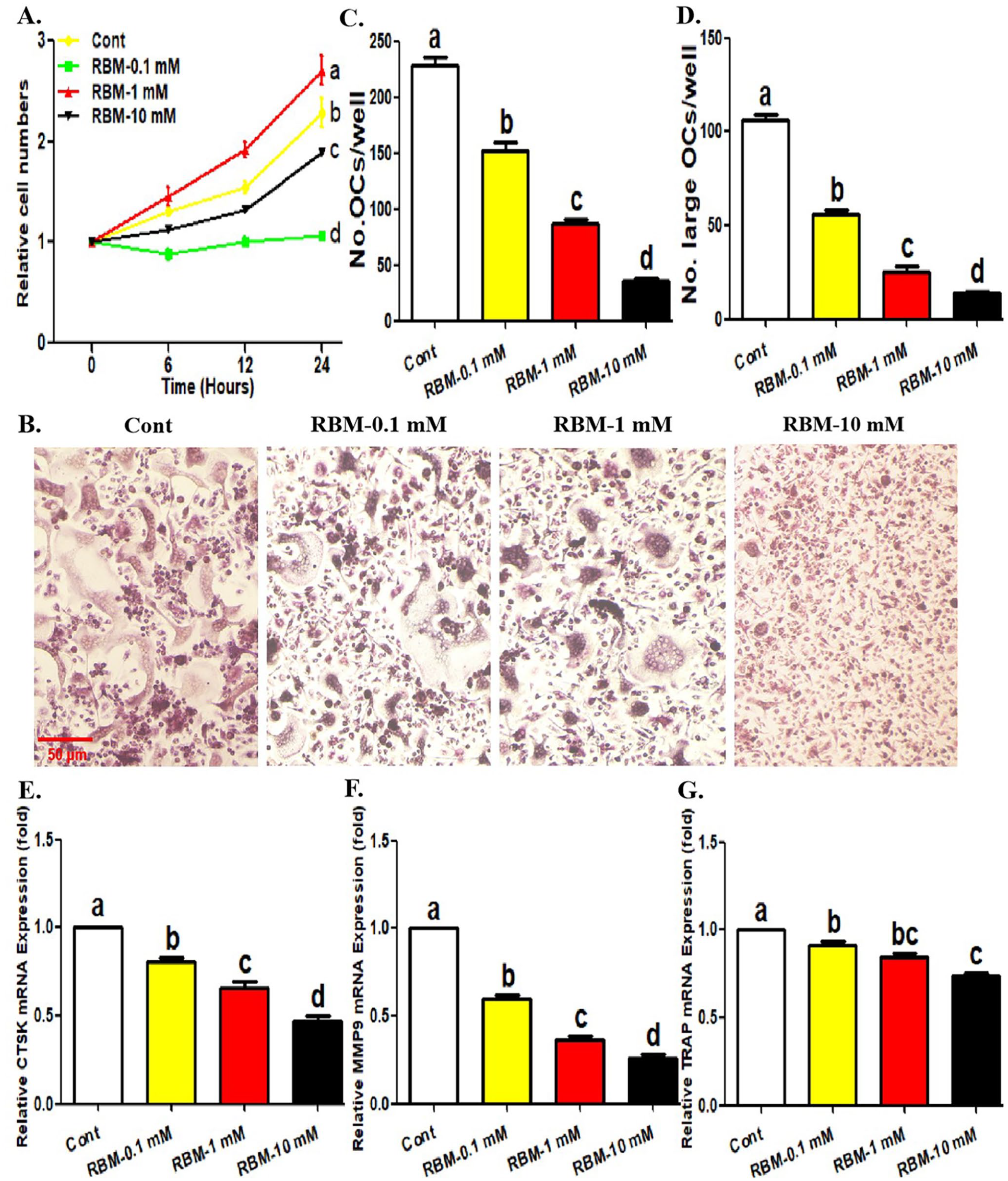
Treatment of RBM inhibits osteoclast differentiation and OCP proliferation. (A) Following the incubation with different concentrations of RBM (0, 0.1, 1, or 10 mM) for indicated times (0, 6, 12, and 24 h) in the presence of RANKL and M-CSF, the proliferative level of OCPs was detected using CCK-8 assays. (B to D) Following the incubation with different concentrations of RBM (0, 0.1, 1, or 10 mM) for 4 days in the presence of RANKL and M-CSF, the formation of mature osteoclasts (TRAP+ multinucleate cells) was evaluated using TRAP staining. The histogram in (C) represents the quantitative results of mature osteoclasts in (B), and the histogram in (D) represents the quantitative results of large osteoclasts over five nuclei in (B). (E to G) Following the incubation with different concentrations of RBM (0, 0.1, 1, or 10 mM) for 4 days in the presence of RANKL and M-CSF, mRNA level of osteoclastic genes was detected by qRT-PCR assays. The data are presented as the mean ± SEM from three independent experiments.
Treatment of RBM inhibited RANKL-related autophagy in OCPs
Next, we observed the effect of RBM on the autophagic activity in OCPs. As shown in Figure 2(A) and (B), RBM reduced the conversion rate of LC3 (shown as the ratio of LC3II and LC3I) in OCPs in a concentration-dependent manner. Also, 10 μM of RBM significantly inhibited the number of autophagosomes/autolysosomes in OCPs (Figure 2(C) and (D)). In addition, the addition of lysosomal protease inhibitor (E64d + Pepstatin A) enhanced LC3 transformation in OCPs with or without RBM, and RBM inhibited LC3 transformation in the presence or absence of E64d + Pepstatin A (Figure 2(E) and (F)), which indicated that autophagic flux is smooth, and this autophagy experimental system is effective.
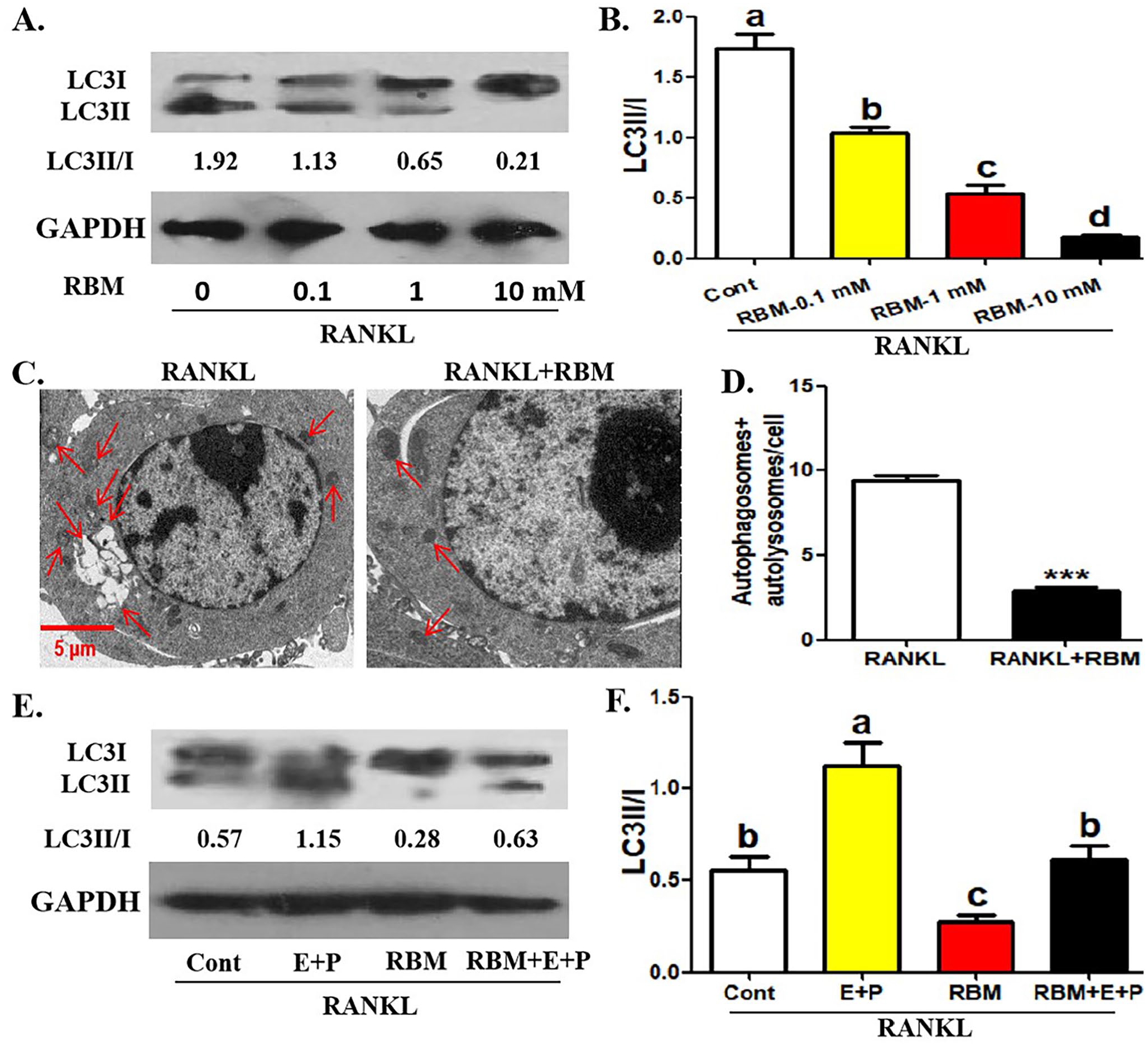
Treatment of RBM inhibits RANKL-related autophagy in OCPs. (A, B) Following the incubation with different concentrations of RBM (0, 0.1, 1, or 10 mM) for 12 h in the presence of RANKL, LC3 protein level in OCPs was detected using Western blotting assays. LC3 conversion rate was defined as the ratio of LC3II/LC3I. (C, D) Following the incubation with RBM (10 mM) for 24 h in the presence of RANKL, the representative images regarding autophagosomes and autolysosomes (red arrows) in OCPs were acquired under TEM. Scale bar, 5 μm. The histogram in (F) represents the quantitative results of autophagosomes + autolysosomes in E (45 cells from three independent experiments). (E, F) Following the incubation with RBM (10 mM) along with or without E64d + Pepstatin A for 12 h in the presence of RANKL, LC3 protein level in OCPs was detected using Western blotting assays. LC3 conversion rate was defined as the ratio of LC3II/LC3I. The data are presented as the mean ± SEM from three independent experiments.
Treatment of TAT-Beclin1 reversed RBM-inhibited osteoclast differentiation and OCP autophagy
Then, we observed the relationship between Beclin1-dependent autophagy and RBM-treated osteoclast formation using Beclin1 pharmacological activator TAT-Beclin1. As shown in Figure 3(A) to (C), the addition of TAT-Beclin1 enhanced BECN1 mRNA expression and Beclin1 protein expression in OCPs with or without RBM, which confirmed the pharmacological effect of TAT-Beclin1. RBM administration inhibited Beclin1 protein expression and BECN1 mRNA expression in OCPs (Figure 3(A) to (C)). In addition, the application of TAT-Beclin1 restored Beclin1 protein expression, LC3 transformation, and LC3-puncta formation in OCPs inhibited by RBM (Figure 3(A) to (F)). Similarly, TAT-Beclin1 application not only significantly promoted the number and size of mature osteoclasts, but also reversed the inhibition of RBM on osteoclastogenic parameters (Figure 3(G) to (I)). These results suggested that Beclin1-dependent autophagy may be involved in RBM-inhibited osteoclast formation.
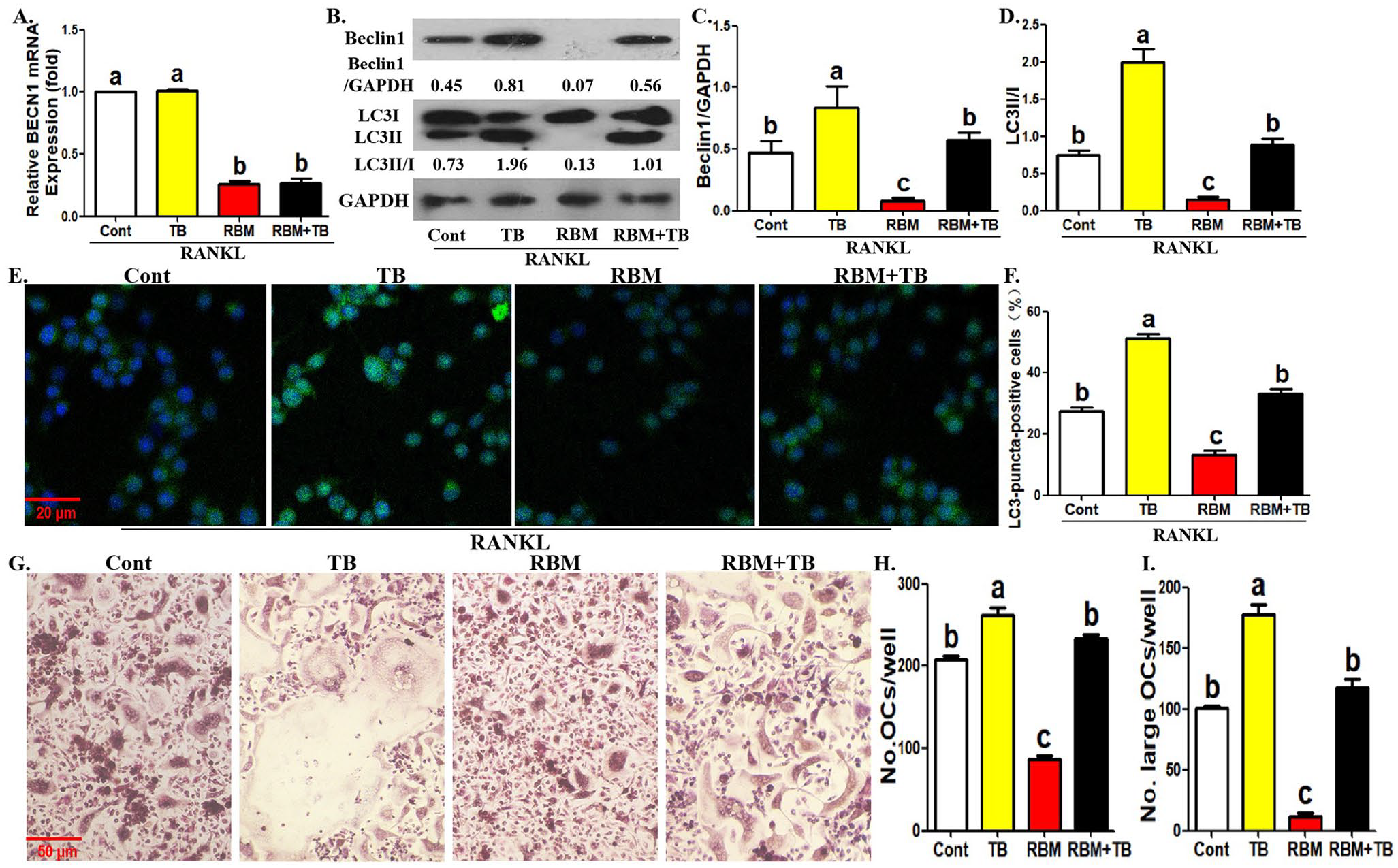
Treatment of TAT-Beclin1 reverses RBM-inhibited osteoclast differentiation and OCP autophagy. (A) Following the incubation with RBM along with or without TAT-Beclin1 (10 μM) for 12 h in the presence of RANKL, BECN1 mRNA level in OCPs was detected by qRT-PCR assays. (B to D) Following the incubation with RBM along with or without TAT-Beclin1 for 12 h in the presence of RANKL, the protein level of Beclin1 and LC3 in OCPs was detected using Western blotting assays. LC3 conversion rate was defined as the ratio of LC3II/LC3I. (E, F) Following the incubation with RBM along with or without TAT-Beclin1 for 12 h in the presence of RANKL, LC3-puncta formation in OCPs was assessed using immunofluorescence staining. The representative images regarding LC3-puncta were acquired via fluorescent microscope. Scale bar, 20 μm. The histogram in (F) represents the quantitative results of LC3-puncta-positive cells in (E) (⩾5 dots, 50 cells per field, n = 5). (G to I) Following the incubation with RBM along with or without TAT-Beclin1 for 4 days in the presence of RANKL and M-CSF, the formation of mature osteoclasts (TRAP+ multinucleate cells) was evaluated using TRAP staining. The histogram in (H) represents the quantitative results of mature osteoclasts in (G), and the histogram in (I) represents the quantitative results of large osteoclasts over five nuclei in (G). The data are presented as the mean ± SEM from three independent experiments.
Treatment of RBM recovered RANKL-induced p65 phosphorylation and nuclear translocation in OCPs
We documented the inhibitory effect of RBM on the protein and gene expression of Beclin1 in OCPs. Previous literature described that RANKL promotes the transcription of BECN1 through NF-κB, thus promoting OCP autophagy. 15 In addition, RBM could inhibit p65 phosphorylation and nuclear translocation in OCPs, thus suppressing RANKL-induced NF-κB pathway. 7 Therefore, the relationship between RBM-inhibited Beclin1 expression and its inhibition on p65 phosphorylation and nuclear translocation deserves further exploration. Accordingly, we investigated the effect of RBM on RANKL-stimulated p65 phosphorylation and nuclear translocation. It was observed that RBM inhibited p65 phosphorylation level in OCPs in a concentration-dependent manner (Figure 4(A) amd (B)). In addition, RANKL caused an increased level of p65 protein in the nucleus and a decreased level of p65 protein in the cytoplasm in OCPs, which was recovered by the addition of RBM (Figure 4(C) to (E)). Under the same conditions, BECN1 mRNA expression and Beclin1 protein expression in OCPs promoted by RANKL were also partially reversed by RBM application (Figure 4(F) to (H)). These results suggested that RBM-inhibited p65 phosphorylation and nuclear translocation may be related to RBM-inhibited Beclin1 expression.
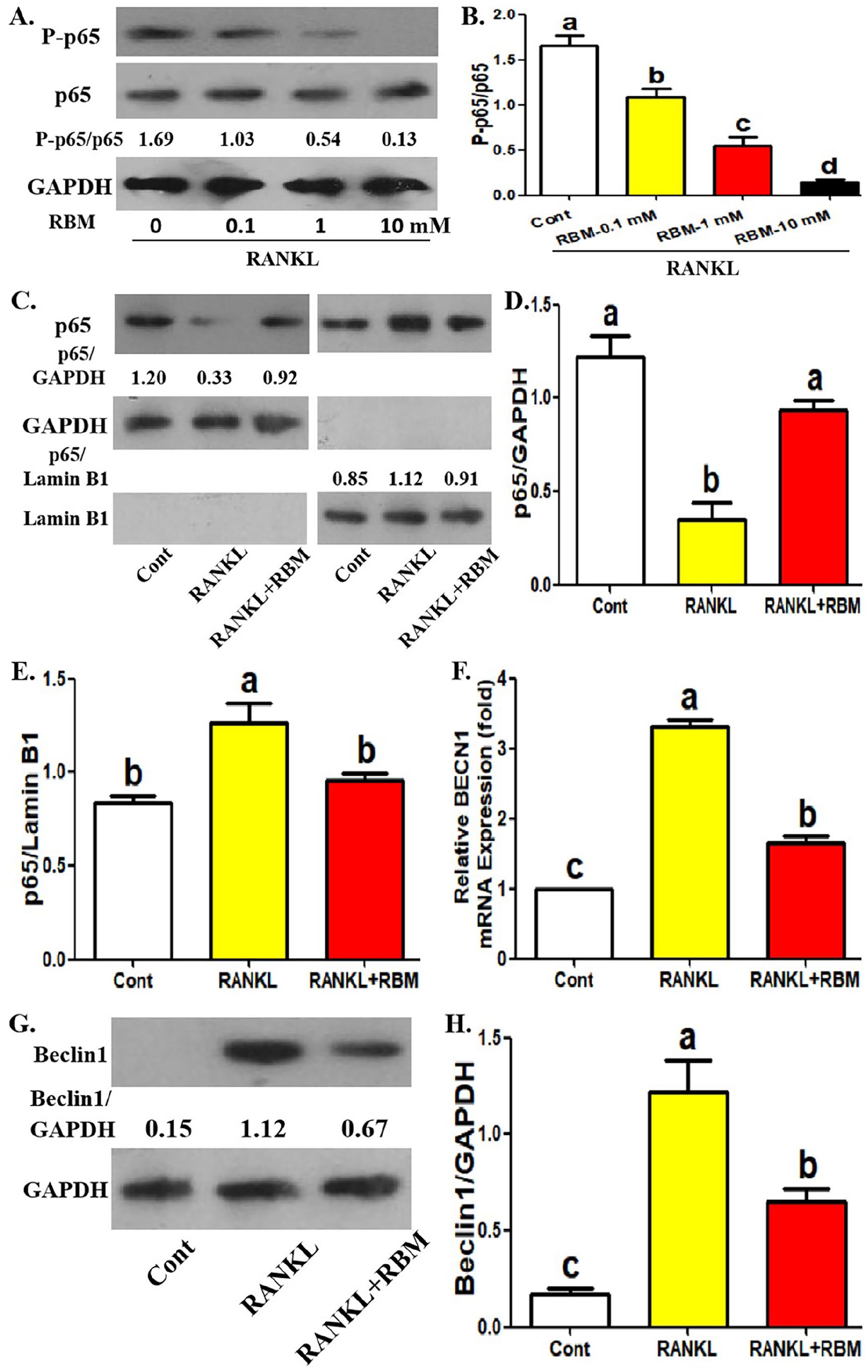
Treatment of RBM recovers RANKL-induced p65 phosphorylation and nuclear translocation in OCPs. (A, B) Following the incubation with different concentrations of RBM (0, 0.1, 1, or 10 mM) for 12 h in the presence of RANKL, P-p65 and p65 protein level in OCPs was detected using Western blotting assays. The relative expression of P-p65 is represented by the ratio of P-p65 to p65. (C to E) Following the incubation with RANKL along with or without RBM for 12 hours, p65 level of separated nuclear protein and separated cytoplasmic protein in OCPs was detected using Western blotting assays. The internal references of nuclear protein and cytoplasmic protein are Lamin B1 and GAPDH, respectively. (F) Following the incubation with RANKL along with or without RBM for 12 h, BECN1 mRNA level in OCPs was detected by qRT-PCR assays. (G, H) Following the incubation with RANKL along with or without RBM for 12 h, Beclin1 protein level in OCPs was detected using Western blotting assays. The data are presented as the mean ± SEM from three independent experiments.
Overexpression of p65 reversed RBM-inhibited osteoclast differentiation and OCP autophagy
To clarify the role of p65 in RBM-regulated OCP autophagy and osteoclast differentiation, we used gene transduction technology to explore the effect of p65 overexpression on RBM efficacy. The efficiency of p65 overexpression was first identified (Figure 5(A)). As shown in Figure 5(B) to (D), BECN1 mRNA level and Beclin1 protein level in OCPs inhibited by RBM were recovered by p65 overexpression. Moreover, the inhibitory effects of RBM on LC3 transformation and LC3-puncta formation in OCPs were blocked by p65 overexpression (Figure 5(C), (E) to (G)). The above results further indicated that p65 is involved in RBM-regulated BECN1 transcription and Beclin1-dependent autophagy in OCPs. In addition, the inhibition of RBM on the number and size of mature osteoclasts was recovered by p65 overexpression (Figure 5(H) to (J)), which suggested that p65 deletion may be involved in RBM’s efficacy on osteoclast formation.
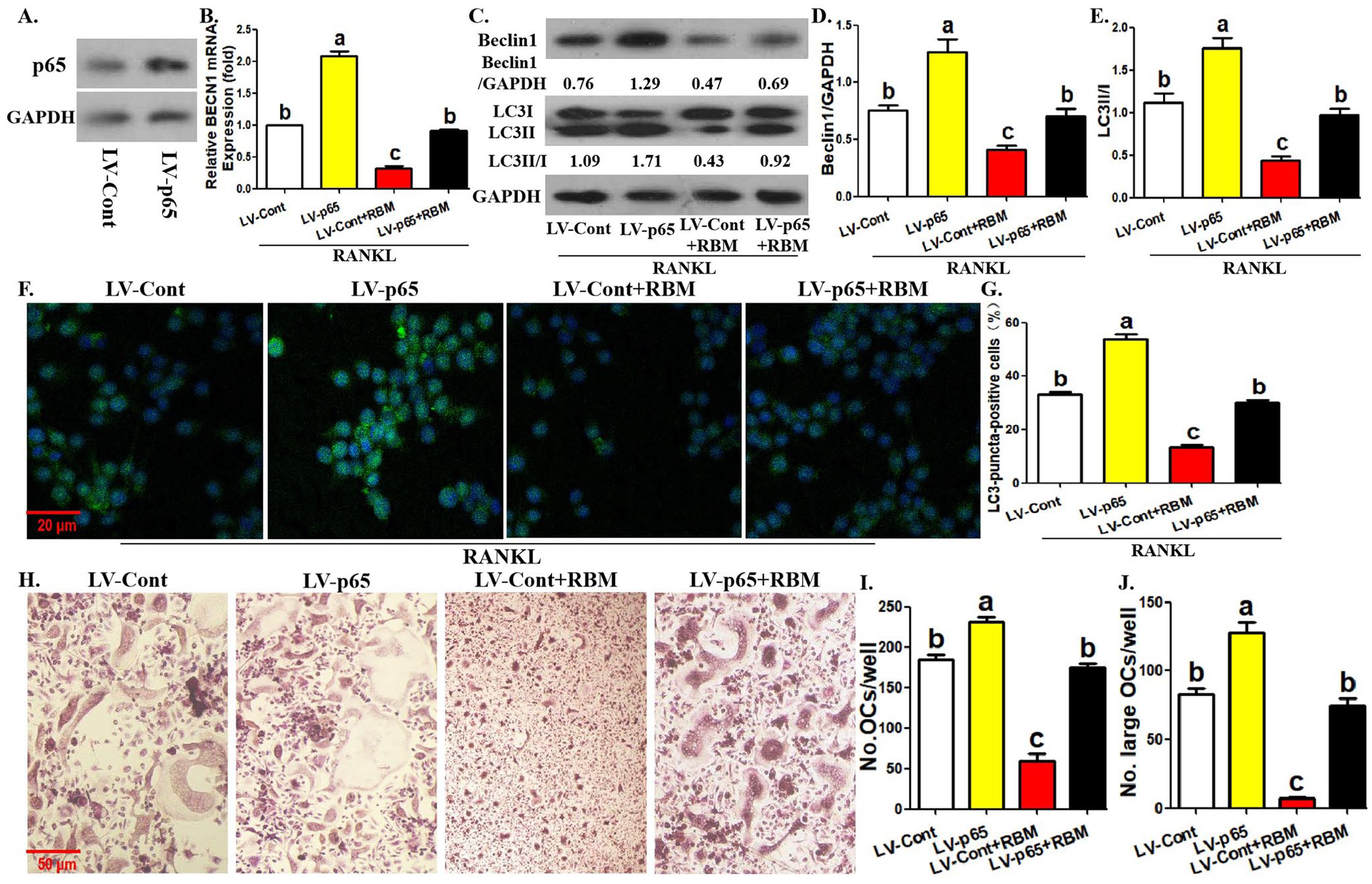
p65 overexpression reverses RBM-inhibited osteoclast differentiation and OCP autophagy. (A) p65 protein level in p65-overexpressing OCPs was detected using Western blotting assays. (B) Following the incubation with RBM for 12 h in the presence of RANKL, BECN1 mRNA level in p65-overexpressing and control OCPs was detected by qRT-PCR assays. (C to E) Following the incubation with RBM for 12 h in the presence of RANKL, the protein level of Beclin1 and LC3 in p65-overexpressing and control OCPs was detected using Western blotting assays. LC3 conversion rate was defined as the ratio of LC3II/LC3I. (F, G) Following the incubation with RBM for 12 h in the presence of RANKL, LC3-puncta formation in p65-overexpressing and control OCPs was assessed using immunofluorescence staining. The representative images regarding LC3-puncta were acquired via fluorescent microscope. Scale bar, 20 μm. The histogram in (G) represents the quantitative results of LC3-puncta-positive cells in (F) (⩾ 5 dots, 50 cells per field, n = 5). (H to J) Following the incubation with RBM for 12 h in the presence of RANKL and M-CSF, the formation of mature osteoclasts (TRAP+ multinucleate cells) from p65-overexpressing and control OCPs was evaluated using TRAP staining. The histogram in (I) represents the quantitative results of mature osteoclasts in (H), and the histogram in (J) represents the quantitative results of large osteoclasts over five nuclei in (H). The data are presented as the mean ± SEM from three independent experiments.
Discussion
As a bioactive substance of M. officinalis How., RBM can be used to treat osteoporosis due to its inhibitory effect on osteoclastogenesis.7,8 Autophagy is of great significance in osteoclastogenesis.11 –14 Therefore, this study explored the relationship between RBM-regulated osteoclastogenesis and autophagic responses. Our experimental data demonstrated for the first time the inhibitory effect of RBM on OCP autophagy during osteoclastogenesis and the underlying mechanism.
First, RBM not only significantly inhibited RANKL-induced OCP autophagy, but also reduced Beclin1 expression level. Previous study showed that M. officinalis oligosaccharides can regulate mitochondrial autophagy in astrocytes. 21 Our results identified the inhibitory effect of another extract from M. officinalis How. on RANKL-induced OCP autophagy. As a key autophagic regulator, Beclin1 regulates autophagic activity relying on the state of its downstream type III phosphoinositol 3-kinase (PI3KC3). 16 Beclin1 and phosphorylated PI3KC3 form the Beclin1-PI3KC3 complex, thereby promoting autophagy. 16 Therefore, we believe that Beclin1-dependent autophagy activation is the key biological basis for RBM-inhibited OCP autophagy and osteoclastogenesis. The pharmacological activator of Beclin1 could recover the inhibitory effects of RBM on osteoclast differentiation and OCP autophagy, which further confirms the above theory. RANKL can promote the transcription of the gene form of Beclin1, BECN1, through the transcription factor NF-κB, thus promoting Beclin1 expression and inducing OCP autophagy. 15 RBM exerts an inhibitory effect on RANKL-induced p65 phosphorylation and p65 nuclear translocation in OCPs. 7 It is well known that p65 is an important component of NF-κB and dominates the transcription characteristics of NF-κB, which left an interesting scientific question: can RBM rely on the inhibition of NF-κB p65 activation to suppress BECN1 transcription and Beclin1-dependent autophagy in OCPs. Our data showed that RBM synchronously inhibited the phosphorylation and nuclear translocation of p65, as well as BECN1 mRNA level and Beclin1 protein level. Moreover, BECN1 mRNA level and Beclin1 protein level in OCPs inhibited by RBM were restored by p65 overexpression. These experimental data confirmed that RBM suppresses the transcription of BECN1 by inhibiting the activation of NF-κB p65, thereby repressing Beclin1 expression level. In addition, the inhibitory effect of RBM on OCP autophagy and osteoclastogenesis was also reversed by p65 overexpression, which further demonstrated that inhibition of BECN1 transcription dominated by NF-κB p65 is the key to RBM-suppressed Beclin1-dependent OCP autophagy, which hinders RANKL-induced osteoclastogenesis. Our work model diagram was shown in Figure 6. Our experimental data are conducive to optimizing the extraction of M. officinalis How. active ingredients and formulating RBM-related treatment strategies in the future. This study was based on in vitro assays, and currently there are no other studies indicating the in vivo model related to our study. In the future, relevant phenomena should be further explored in vivo. In addition, the potential mechanism of RBM-inhibited OCP autophagy was only partially elucidated in this study, and further researches are needed to broaden and deepen the regulatory mechanism of RBM on OCP autophagy.
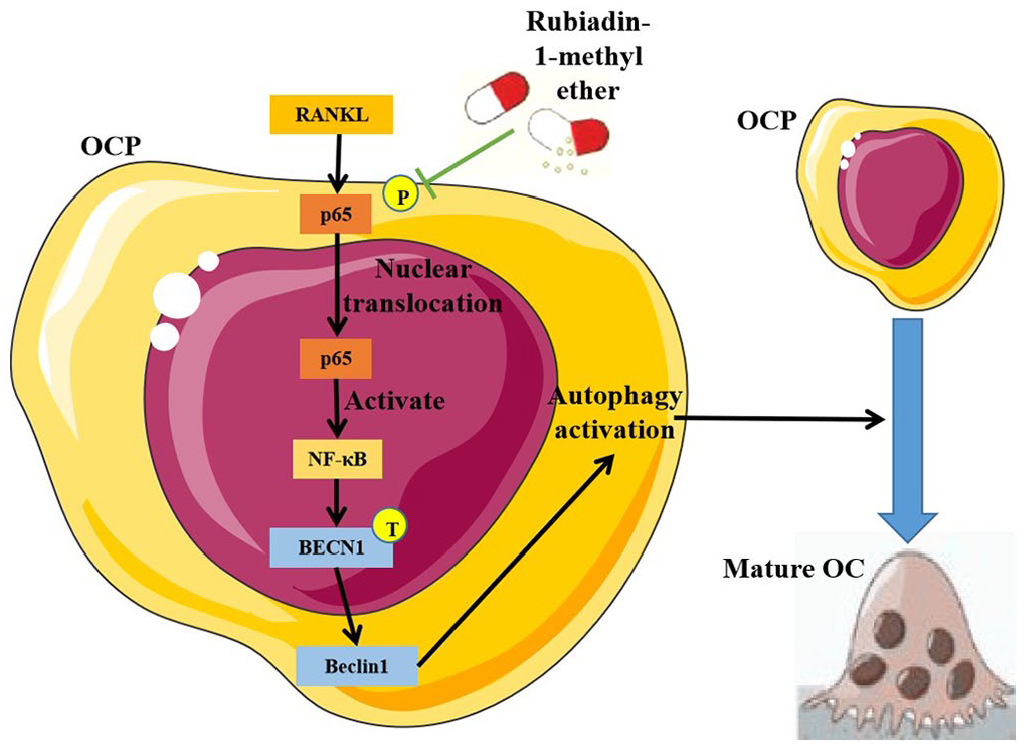
The working model diagram of this study.
In summary, our study elucidated the inhibitory effect of RBM on OCP autophagy and the significance of Beclin1-dependent autophagy for RBM-inhibited osteoclastogenesis. Importantly, the above data described a biological process: RBM suppresses RANKL-induced transcription of autophagy gene BECN1 by inhibiting the activation of NF-κB p65, thereby suppressing Beclin1 expression and Beclin1-dependent autophagy activation, which results in the inhibited osteoclastogenesis. Our study not only describes for the first time the molecular mechanism of RBM-inhibited osteoclastogenesis from the perspective of autophagy, but also provides more potential clues for the treatment of RBM in osteoclastic bone loss.
Footnotes
Authors’ Contributions
HW and SC conceived and designed experiments; SC, YC, and JC performed experiments, analyzed data, prepared figures, and helped with writing of the manuscript; WW and JP helped with experimental operation and data analysis; HW and SC wrote the manuscript. HW reviewed and edited this manuscript. All authors have read and agreed to the published version of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Fujian Provincial Health Commission Young and middle-aged backbone talent training project (2020GGA064).