
Abstract
Glucagon-like peptide-1 (GLP-1), secreted by intestinal L-cells, plays a pivotal role in the modulation of β-cell insulin secretion in a glucose-dependent manner, concurrently promoting β-cell survival and β-cell mass. Notably, GLP-1 has emerged as an effective second-line treatment for type 2 diabetes mellitus, gaining further prominence for its pronounced impact on body weight reduction, positioning it as a potent antiobesity agent. However, the mechanism by which GLP-1 improves obesity remains unclear. Some reports suggest that this mechanism may be associated with the regulation of adipokine synthesis within adipose tissue. Chemerin, a multifunctional adipokine and chemokine, has been identified as a pivotal player in adipocyte differentiation and the propagation of systemic inflammation, a hallmark of obesity. This review provides a comprehensive overview of the mechanisms by which GLP-1 and chemerin play crucial roles in obesity and obesity-related diseases. It discusses well-established aspects, such as their effects on food intake and glycolipid metabolism, as well as recent insights, including their influence on macrophage polarization and adipose tissue thermogenesis. GLP-1 has been shown to increase the population of anti-inflammatory M2 macrophages, promote brown adipose tissue thermogenesis, and induce the browning of white adipose tissue. In contrast, chemerin exhibits opposite effects in these processes. In addition, recent research findings have demonstrated the promising potential of GLP-1-based therapies in directly or indirectly regulating chemerin expression. In an intriguing reciprocal relationship, chemerin has also been newly identified as a negative regulator of GLP-1 in vivo. This review delineates the intricate interplay between GLP-1 and chemerin, unraveling their mutual inhibitory interactions. To the best of our knowledge, no previous reviews have focused on this specific topic, making this review particularly valuable in expanding our understanding of the endocrine mechanisms of obesity and providing potential strategies for the treatment of obesity and related diseases.
Impact Statement
This review provides a comprehensive analysis of the pivotal role of glucagon-like peptide-1 and chemerin in the treatment of obesity. GLP-1 exhibits potent insulinotropic effects and plays a crucial role in the regulation of glucose homeostasis, making it an attractive therapeutic target for obesity management. Conversely, chemerin, an emerging adipokine and inflammatory factor, plays a critical role in adipogenesis and contributes to metabolic dysregulation associated with obesity. Notably, emerging research has highlighted a potential interaction between GLP-1 and chemerin, suggesting their intertwined roles in modulating adipose tissue functions, such as macrophage polarization and thermogenesis. By shedding light on the reciprocal inhibition between GLP-1 and chemerin, this comprehensive review enhances our understanding of the endocrine mechanisms underlying obesity. Importantly, this review is the first to comprehensively address this topic, filling a significant gap in the existing literature and establishing a foundation for future research and therapeutic interventions in the field of obesity.
Introduction
Obesity, a widespread global epidemic, significantly contributes to the development of numerous obesity-related conditions, including metabolic syndrome, type 2 diabetes mellitus (T2DM), dyslipidemia, fatty liver disease, hypertension, cardiovascular disease, and specific cancers, thereby leading to diminished quality of life and reduced life expectancy. 1 Glucagon-like peptide-1 (GLP-1), one of the main peptides of the incretin family, is secreted by intestinal L-cells induced by feeding stimulation after a meal. The most well-known action of GLP-1 is to potentiate postprandial insulin release in a glucose-dependent manner through directly acting on pancreatic β-cells, which is essential for glucose homeostasis. 2 In addition, exogenous infusion of GLP-1 has been shown to lower glucagon secretion, inhibit hepatic glucose production, delay gastric emptying, and induce satiety, which collectively leads to improved glycemia and enhanced insulin sensitivity. The insulinotropic and anorexigenic actions of GLP-1 make it an appealing candidate for the treatment of obesity. 3
However, the mechanism by which GLP-1 improves obesity remains unclear. Some reports suggest that this mechanism may be associated with the regulation of adipokine synthesis within adipose tissue.4,5 Chemerin, an adipokine with inflammatory properties, highly expressed in adipose tissue and liver, can regulate adipogenesis and adipocyte metabolism and is noted to be an important factor involved in the inflammatory process cascade. 6 At present, the majority of known chemerin functions predominantly operate through the mediation of chemokine-like receptor 1 (CMKLR1). 7 In recent years, a large number of studies, including our previous study, 8 have shown that chemerin/CMKLR1 is closely related to obesity and obesity-related disorders of glucose and lipid metabolism.9,10
In addition, the exploration of the relationship between GLP-1 and chemerin has aroused more research interest recently, and it has been reported in 2015 at first that the increases of GLP-1 by glucagon-like peptide-1 receptor (GLP-1R) agonists could reduce chemerin expression both in the liver and circulation of rats, accompanied by obvious improvement of insulin sensitivity. 11 In an intriguing reciprocal relationship, chemerin has also been newly identified as a negative regulator of GLP-1 in vivo. 12 Considering the multifaceted beneficial effects of GLP-1 on adipose tissue during obesity, its regulation of the adipokine chemerin opens up new avenues for exploring relevant mechanisms. Furthermore, as an inflammatory factor that can act on colonic tissue, chemerin also offers a novel pathway for investigating the influences of obesity on colonic GLP-1 secretion.
After briefly introducing the metabolic effects of these two molecules, this review primarily delves into the novel mechanisms by which GLP-1 and chemerin influence adipose tissue (specifically, macrophage polarization and thermogenesis) within the context of obesity. In addition, this review delineates the intricate interplay between GLP-1 and chemerin, unraveling their mutual inhibitory interactions. To the best of our knowledge, no previous reviews have focused on this specific topic, making this review particularly valuable in expanding our understanding of the endocrine mechanisms of obesity and providing potential strategies for the treatment of obesity and related diseases.
What is known about the metabolic effects of GLP-1 and chemerin
GLP-1 promotes insulin secretion, reduces hepatic steatosis, inhibits appetite, and improves osteogenesis
As mentioned in section “Introduction,” the insulinotropic and anorexigenic actions are the main effects of GLP-1. For insulinotropic action, agonists of the GLP-1R also improve glycemic control via chronic action to preserve β-cell mass through stimulating proliferation and inhibiting apoptosis of β-cell and suppressing glucagon secretion of α-cell. 13 GLP-1 inhibition of glucagon secretion has been demonstrated in vivo in numerous species including mice and humans, as well as in isolated intact murine islets. 13 In addition, a randomized trial in humans indicated that treatment over one year with GLP-1R agonists liraglutide as an adjunct to diet and exercise was associated with clinically meaningful weight loss in overweight or obese patients, with concurrent reductions in glycemic variables. 14 Liraglutide has been approved for weight control in overweight and obese adults and has become a recommended drug for obesity treatment. 15
As an effective factor that inhibits food intake, GLP-1 can induce satiety and plays an important role in regulating appetite.16,17 Hypothalamus is the main regulator of thirst and hunger and receives signals from the nucleus of solitary tract. 18 GLP-1-expressing neurons in the hindbrain send robust projections to the paraventricular nucleus of the hypothalamus (PVN), which is one of the several brain centers implicated in food intake behavior. 19 In central nervous system (CNS), GLP-1, a neurotransmitter in a brain stem-hypothalamic pathway, mediates synaptic plasticity via protein kinase A (PKA) pathway to augment excitatory neurotransmission in PVN to suppress food intake. 19 Not only PVN, a report recently showed that GLP-1R expressing neurons in the dorsomedial hypothalamic nucleus (DMH) may mediate the suppression of food intake. The deletion of GLP-1R-positive neurons in DMH induced hyperphagia, disruption of diurnal feeding patterns, and obesity. 20
In addition, exogenous GLP-1 has been shown to exert beneficial effects in bone, liver, and muscle. Most studies have reported that GLP-1 might improve bone growth and bone remodeling by GLP-1R. For instance, healthy obese women often had a loss of bone mass after weight loss, and treatment with liraglutide made them have less loss of bone mineral content and bone markers. 21 In a rat bone loss model, GLP-1R agonists promoted bone formation and increased bone mass with an increase in osteoblast number and serum bone formation markers, while the adipocyte number was decreased. 22 The promoting effect of GLP-1 on osteogenesis is achieved by regulating the differentiation of bone mesenchymal stem cells preferably to osteoblasts rather than adipocytes. 22 Besides, GLP-1R agonists could reduce hepatic steatosis, decrease liver inflammation, and attenuate hepatocyte injury in preclinical models of non-alcoholic steatohepatitis, but the mechanistic understanding of how GLP-1R agonists attenuate non-alcoholic steatohepatitis remains unclear. 16 Moreover, GLP-1 acutely recruited muscle microvasculature, increased muscle delivery of insulin, enhanced muscle use of glucose, and improved insulin’s metabolic action in the insulin-resistant states. 16
Chemerin promotes adipocyte differentiation, regulates glucose homeostasis and appetite, and inhibits osteoblastogenesis
As a chemoattractant protein, chemerin promotes chemotaxis of immature dendritic cells and macrophages, which was identified as a new adipokine in 2007. 23 Chemerin secretion from mature adipocytes exerts a proadipogenic drive, acting as a paracrine/autocrine factor to promote adipocyte differentiation through the activation of CMKLR1, thereby contributing to the expansion of adipose mass. 7 Interestingly, at normal concentrations, chemerin plays a crucial role in adipogenesis, lipolysis, and maintaining blood glucose homeostasis. However, excess chemerin due to increased fat mass is likely to cause inflammation in adipose tissue, glycolipid metabolism disorders, and insulin resistance (IR) in multiple organs, closely associated with the development of obesity. 24 In addition, chemerin has been implicated in the regulation of glucose homeostasis, although conflicting research findings have emerged. This discrepancy could potentially be attributed to the existence of multiple receptors for chemerin. Apart from CMKLR1, chemerin also binds to two other distinct receptors: G protein-coupled receptor 1 (GPR1) and a non-signaling seven-transmembrane domain receptor known as CC-motif chemokine receptor-like 2 (CCRL2).7,25 Investigations involving knockout models of CMKLR1, encompassing both whole-body and arcuate nucleus-specific approaches, have unveiled minimal effects on glucose homeostasis.26–28 Conversely, mice lacking the GPR1 receptor have displayed heightened glucose intolerance. 29 These findings suggest that chemerin’s regulation of glucose homeostasis may be mediated by GPR1.
Chemerin/CMKLR1 has been the subject of contradictory reports regarding its acute effects on food intake. Rats that received acute injections of chemerin 30 or a CMKLR1 antagonist 28 into the hypothalamus showed no significant effect on food intake after 24 h. On the contrary, acute intravenous injection of chemerin in rats resulted in a significant reduction in food intake after 24 h. 31 In addition, the chronic effects of chemerin/CMKLR1 on food intake are controversial.28,31,32 To gain a comprehensive understanding of the effects of chemerin/CMKLR1 on food intake, future investigations should focus on specific animal strains, disease models, administration sites, dosages, and intervention durations. From a mechanistic perspective, there is evidence suggesting that the long-term impact of chemerin on food intake and body weight is not directly or simply correlated with the known appetite and energy balance pathways, but it may involve other hypothalamic mechanisms. 31 In the hypothalamus of rats, chemerin and CMKLR1 transcripts are localized in the ependymal cells and tanycytes, which are specialized glial cells lining the third ventricle and extending into the arcuate nucleus, closely associated with homeostatic appetite regulation. The regulatory effect of chemerin on body weight and appetite might be mediated by hypothalamic remodeling, for long-term central chemerin infusion resulting in morphological changes of the hypothalamus by increasing vimentin (a marker for visualizing cells of glial origin such as tanycytes and ependymal cells) immunolabeling of tanycytes. 31 These findings provide valuable insights into the complex mechanisms underlying appetite control.
Besides, chemerin inhibits osteoblastogenesis in marrow when it exerts a positive stimulus to adipogenesis. 33 Osteoblasts and adipocytes of bone marrow are remote descendants of the same mesenchymal stromal cell; thus, a preferential differentiation into adipocytes cells may be at the expense of osteoblasts decrease. 33 Chemerin promotes adipogenic differentiation, consequently, often inhibits osteogenic differentiation of bone mesenchymal stem cells, which may eventually affect bone mass. This may be a reason why obesity and obesity-related diseases are often accompanied by osteoporosis. 34 A study based on a large sample size showed that chemerin had an inverse association with bone mass and a positive relation with medium/high risk of osteoporotic fractures in obese men and women. 35 In conclusion, both GLP-1 and chemerin have been reported to exert contrary effects on metabolisms of multiple organs including brain, pancreas, muscle, liver, and bone (Figure 1).
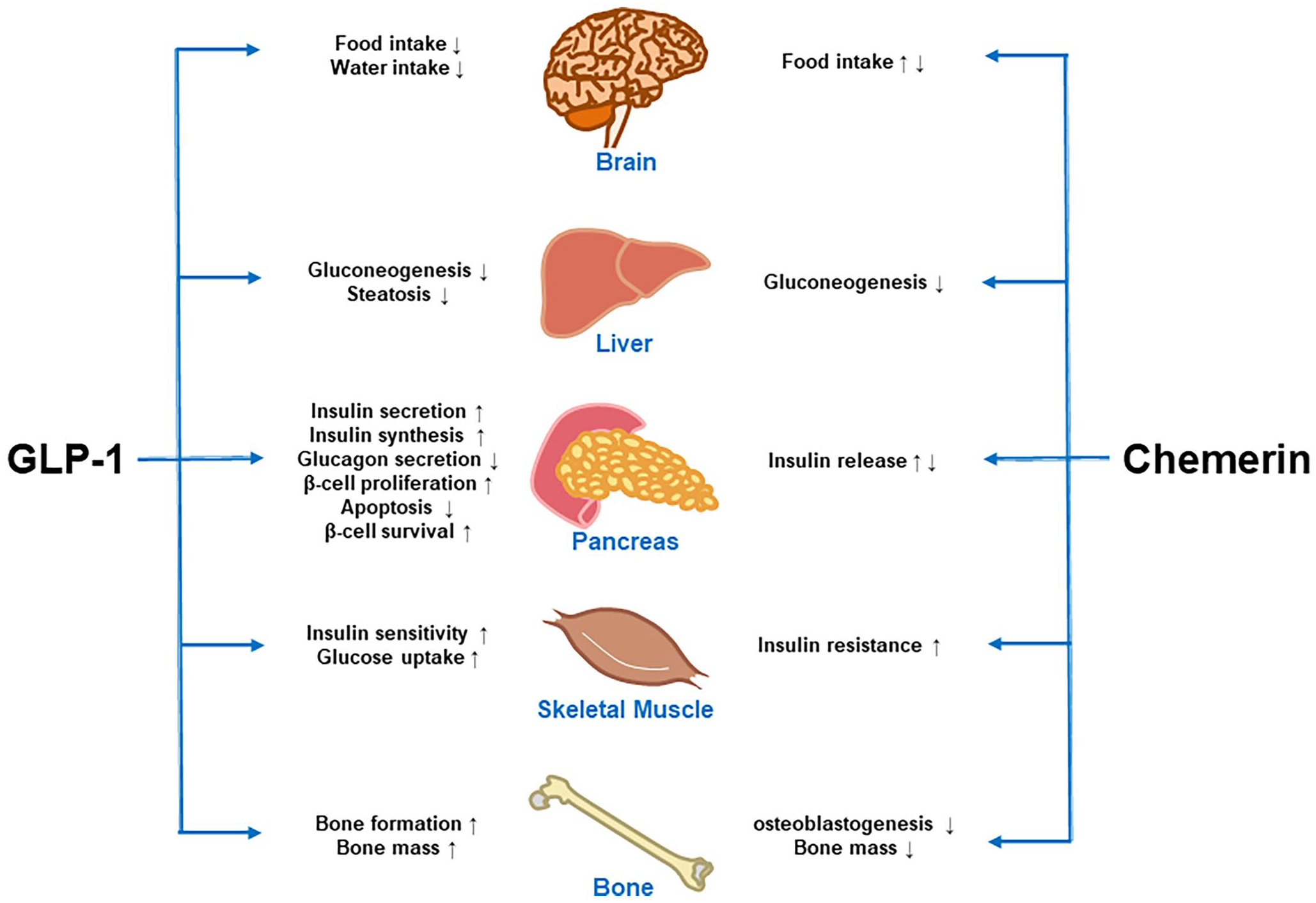
Summary of the known contrary effects of GLP-1 and chemerin on the metabolisms of multiple organs.
New insights of GLP-1 and chemerin on macrophage polarization of adipose tissue
Recently, there has been increasing attention on treatment and prevention strategies for weight loss and obesity-induced metabolic disorders. Macrophages are now considered ideal therapeutic targets for metabolic diseases due to their phenotypic flexibility. 36 The differentiation of monocytes in the blood into classically activated macrophages (M1) or alternatively activated macrophages (M2) upon stimulation plays a crucial role in adipose tissue function and metabolic health. It is important to clarify that M1 macrophages secrete inflammatory factors like tumor necrosis factor (TNF)-α, interleukin (IL)-6, and IL-1β, which can inhibit insulin receptor serine phosphorylation on adipocytes via c-Jun-N-terminal kinase (JNK), leading to IR and adipose tissue expansion. Conversely, M2 macrophages inhibit inflammatory responses mediated by M1 macrophages and contribute to adipocyte insulin sensitivity by producing IL-10. 37 IL-4, an adaptive cytokine within the immune system, is secreted by activated T-cells. It can facilitate the phosphorylation of signal transducer and activator of transcription (STAT) 6, thereby further activating the initiation of the M2 transcriptional program.38,39 Obesity promotes the polarization of resident macrophages toward the M1 phenotype, leading to a shift in the macrophage population from M2 to M1. This shift further increases pro-inflammatory cytokines and chemokines, inducing chronic inflammation and exacerbating IR. 40 To address adipose tissue macrophage–related inflammation and improve adipose tissue function in obesity, potential macrophage-specific therapeutics could focus on decreasing pro-inflammatory M1 macrophages or increasing anti-inflammatory M2 macrophages. 37
GLP-1 inhibits the polarization of adipose tissue macrophages to pro-inflammatory M1
TNF-α, IL-6, and IL-1β are pro-inflammatory cytokines commonly associated with the M1 phenotype. GLP-1 treatment was found to reduce the release of these inflammatory cytokines in lipopolysaccharide-stimulated macrophages with improved insulin-stimulated glucose uptake. 41 In vivo studies using diet-induced obese (DIO) mice demonstrated that a four-week administration of a GLP-1R agonist significantly reduced the elevated levels of IL-6 and IL-1β in serum and adipose tissue. 42 This implies GLP-1 suppresses the pro-inflammatory activation of macrophages to M1 type in adipose tissue. In addition, obese mice treated with GLP-1 agonist exhibited significant weight loss, reduced fat mass, and alleviated hypoxia in the adipose tissue, which may contribute to the reduced inflammatory mediators and the improved insulin sensitivity in adipose tissue. 42 Some more direct evidence has disclosed that GLP-1 could advance macrophage polarization toward anti-inflammatory M2 phenotype. The results of flow cytometry and real-time polymerase chain reaction (PCR) showed a significant reduction in the number of M1 macrophages and an increase in the number of M2 macrophages in the adipose tissue of obese mice treated with GLP-1 analogue. 36 (Ex-4)2-Fc, an effective GLP-1 analogue treatment, efficiently decreased the obesity-induced secretion of IL-1β, TNF-α, and IL-6, leading to profound amelioration of inflammation in the adipose tissues of DIO mice. 43 However, it should be noted that while (Ex-4)2-Fc reduced the weight of DIO mice by 20%, the percentage of weight loss in ob/ob and db/db mice was only 10%. Moreover, (Ex-4)2-Fc treatment did not significantly alter cytokine expression in the adipose tissue of ob/ob and db/db mice. 43 These findings suggest that the observed reduction in inflammatory cytokines following (Ex-4)2-Fc treatment is associated with weight change. However, further research is needed to fully understand the underlying mechanisms of GLP-1’s effects on cytokine regulation and its relation to weight loss.
As for the mechanisms of GLP-1 affecting the polarization of macrophages, a study in vitro elucidated that the effects of GLP-1 on the increases of M2-specific genes expression and decreases of M1-specific gene expression were fulfilled by the mediation of the GLP-1/JNK/STAT3 signaling pathway. 40 JNK is an important branch of mitogen-activated protein kinase signaling pathways in cells, and activation of JNK leads to the M1 polarization of macrophages and tissue inflammation, 44 which is required for systemic IR caused by diet-induced obesity. 45 STAT, a unique protein family that binds to DNA, usually responds to various extracellular cytokine and growth factor signals, drives events as varied as hematopoiesis, immune fitness, inflammation, tissue repair, adipogenesis, and apoptosis. 46 STAT3 is a repressor protein of inflammatory response, which inhibits inflammation and M1 activation as well as promotes M2 activation. 47 GLP-1/GLP-1R was reported to attenuate the phosphorylation of JNK and its signal transduction through cyclic adenosine monophosphate (cAMP)/PKA signaling pathway, and the suppression of JNK phosphorylation in macrophages upregulated the intracellular levels of phosphorylated STAT3, which served a crucial role in M2 polarization and mediated the anti-inflammation response 40 (Figure 2(A)).
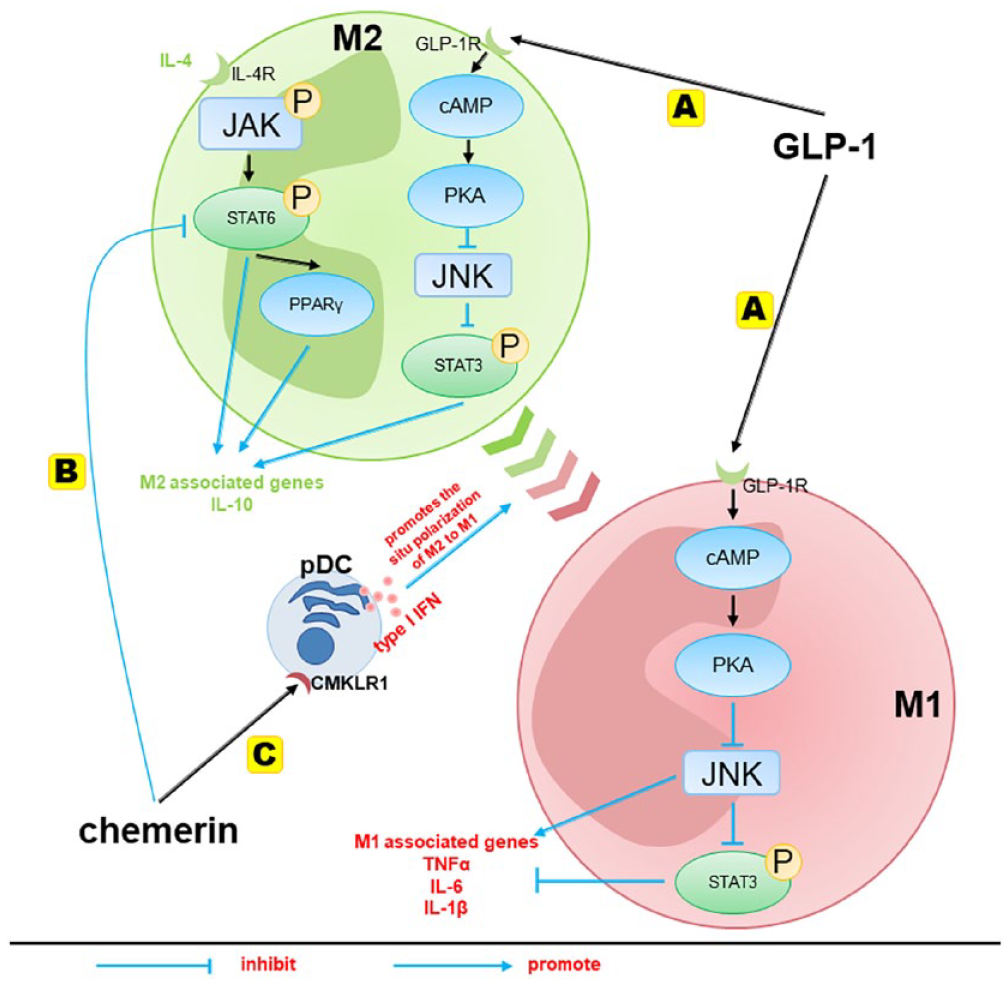
The regulation mechanisms of GLP-1 and chemerin on macrophage polarization. (A) The effect of GLP-1 on macrophage activation, which contributed to M2 polarization and secretion of anti-inflammatory factors by modulating the JNK/STAT3 signaling pathway. (B) Chemerin affects the polarization of macrophages by directly suppressing M2 macrophage polarization through inhibiting the IL-4/STAT6-PPARγ pathway. (C) Chemerin recruits pDCs to secrete type I IFN, thereby promoting the in situ polarization of macrophages from the M2 to the M1 phenotype.
Chemerin inhibits the polarization of adipose tissue macrophages to anti-inflammatory M2
It has been reported in animal studies that weight loss and improved perivascular fat tissue dysfunction in DIO mice were associated with decreased M1 macrophages and lower levels of chemerin. 48 In addition, a recent study showed that electroacupuncture reduced body weight, adiposity, and fat accumulation in DIO mice. This was accompanied by an increased ratio of M2 to M1 macrophages in adipose tissue and a reduction in chemerin secretion. 49 In vitro studies provided direct evidence that chemerin can suppress the polarization of M2 macrophages, as the addition of chemerin reduced the expression of M2-associated genes in IL-4-stimulated macrophages. 50 Notably, chemerin knockout (KO) mice exhibited higher numbers of M2 macrophages in adipose tissue compared to wild-type (WT) mice, suggesting an inhibitory role of chemerin on M2 macrophages. 51 Similarly, CMKLR1 KO mice, which lack the receptor for chemerin, also showed increased M2 macrophage abundance in adipose tissue. After the chronic high-fat diet (HFD) challenge, CMKLR1 KO mice had less weight gain, significantly reduced fat mass, and obvious decreases in inflammatory genes such as TNF-α, IL-6, and IL-1β. 51 These findings collectively demonstrate the potential role of chemerin in inhibiting macrophage polarization to M2 and its impact on adipose tissue inflammation and weight loss.
As for the mechanisms of chemerin affecting the polarization of macrophages, it has been reported that chemerin was able to directly modulate macrophage polarization in vitro by influencing the IL-4/STAT6-dependent mechanism. The T-helper 2 cell-derived IL-4 has a key role in orchestrating M2 activation of macrophages. Macrophages treated by IL-4 share many of the features characteristic of M2-polarized macrophages. 52 It is known that IL-4-induced M2 macrophage polarization is dependent on STAT6, a master regulator of M2 genes. The downstream of the IL-4 signaling involves the activation of various JNK, which leads to the phosphorylation of STAT6.38,39 In addition, STAT6 also induces the expression of transcription factor peroxisome proliferator–activated receptor (PPAR)γ, which acts in synergy with STAT6 to regulate the expression of M2-specific genes and macrophage polarization in obese mice. 50 In vitro, the addition of chemerin significantly reduced the mRNA expression of all the M2 macrophage-associated genes in IL-4-stimulated macrophages and inhibited phosphorylated STAT6 50 (Figure 2(B)).
Furthermore, chemerin not only suppresses M2 macrophage polarization but also plays a role in promoting the in situ polarization of M2 macrophages to M1 macrophages. CMKLR1 is expressed in numerous immune cells that accumulate in obese adipose tissue, including plasmacytoid dendritic cells (pDCs), myeloid dendritic cells, macrophages, and natural killer cells. 6 As a chemotactic protein, chemerin recruits pDCs or macrophages via its functional receptor CMKLR1, 51 and circulating pDCs infiltrated visceral adipose tissue in response to CMKLR1 triggering were the major producers of type I interferons (IFNs) in the body. 53 The related experiment in vitro has shown that type I IFN can fuel the in situ polarization of M2 macrophages to pro-inflammatory M1 macrophages, leading to the propagation of chronic inflammation and the development of IR in visceral adipose tissue 53 (Figure 2(C)). This indicates that the homing and recruitment of pDCs by the chemerin/CMKLR1 axis is the beginning of the macrophage polarization process. Results from CMKLR1 KO mice also support the impairments in numbers of pDCs as well as in their migratory response and homing to chemerin, accompanied by reduced atherosclerosis and increased macrophages M2 at the lesion in CMKLR1 KO mice. 54 So, chemerin affects the polarization of macrophages through: (1) directly suppressing M2 macrophage polarization by inhibiting the IL-4/STAT6-PPARγ pathway and (2) recruiting pDCs to secrete type I IFN, which fuels the in situ polarization of M2 macrophages to pro-inflammatory M1 macrophages.
New insights of GLP-1 and chemerin on thermogenesis of adipose tissue
Brown adipose tissue (BAT) is a key site of heat production (thermogenesis) in mammals that has for many decades been considered an attractive target to induce weight loss. Brown adipocytes result in non-trembling thermogenesis by expressing uncoupling protein-1 (UCP-1), which is a thermogenic protein found in BAT cells and plays an important role in cold and diet-induced thermogenesis. Moreover, clusters of UCP-1 expressing adipocytes with thermogenic capacity also develop in white adipose tissue (WAT) in response to various stimuli, and these adipocytes are named as beige fat cells, which have similar morphology and thermogenic function as BAT. 55 Beige cells in mouse WAT are defined by their multilocular lipid droplet morphology, high mitochondrial content, and the expression of a core set of BAT-specific genes like UCP-1, irisin, and PPARγ co-activator 1-alpha (PGC1-α). 56
The biomedical interest in brown and beige adipocytes has centered on the capacity of these cell types to counteract metabolic diseases, including obesity and T2DM. The reduction of brown adipocytes made mice more susceptible to obesity and metabolic dysfunction, and BAT transplantation improved systemic energy metabolism, glucose homeostasis, and insulin sensitivity. 57 The recruitment and activation of brown and beige adipocytes were able to increase energy expenditure, reduce body weight, and increase insulin sensitivity, whereas UCP-1 deficient mice gained more body weight than WT controls. 58 Therefore, activating the conversion of WAT to beige fat and increasing the activities of brown fat and beige fat or both holds tremendous promise for the treatment of obesity and its related metabolic diseases.
GLP-1 promotes thermogenesis
It is reported that the beneficial effect of GLP-1R agonists on obese mice is associated with the brown remodeling of WAT and the increase of thermogenesis. 59 Chronic peripheral treatment with the novel GLP-1R agonist upregulated the expression of UCP-1 in inguinal WAT of obese mice, suggesting an increase in beige adipocytes upon GLP-1R agonist treatment. 59 As well, GLP-1R agonist also induced brown adipocyte differentiation in skeletal muscle of diabetic mice, including a significant increase in the mRNA levels of thermogenic genes such as UCP-1, β3-adrenergic receptor, and PPARα (a brown fat marker). 58 During the exposure to cold, the body temperatures of untreated obese mice dropped significantly, displaying impaired thermogenesis compared with the GLP-1R agonist treated mice, which kept higher rectal temperature throughout the experiment, suggesting that GLP-1R agonist was able to increase body adaptation to cold exposure by generating more heat. 59 The thermogenesis from beige adipocytes can contribute substantially to energy expenditure, 60 thus inducing the improvements of obesity and IR in mice with diet-induced obesity or obesity-related diseases. In addition, in vitro, treatment of differentiated C2C12 myoblasts with GLP-1R agonist reduced lipid accumulation and triglyceride content, accompanied by significantly upregulated UCP-1. 61 These results indicated that GLP-1/GLP-1R increased brown remodeling of WAT in adipose tissue and skeletal muscle, then increased thermogenesis to alleviate obesity.
As for the mechanisms of GLP-1 affecting thermogenesis, it has been reported that GLP-1 regulated BAT thermogenesis and glycemia clearance via the CNS.62–64 Intracerebroventricular infusion of GLP-1R agonist in both lean and obese mice increased sympathetic outflow toward BAT, resulting in increased thermogenesis as evidenced by increased UCP-1 protein levels and decreased lipid content, and BAT activation may be a major contributor to glucose- and triglyceride-lowering effects as well as body weight loss of central infusion of GLP-1R agonism. 65
Within the CNS, GLP-1R is expressed in numerous neuronal populations, highly expressed in the hypothalamus, 66 and especially highly expressed in the anterior and ventral part of the DMH, which is a key part of the regulation of energy balance and the sympathetic control network and is densely innervated by GLP-1 fibers. 20 DMH neurons send monosynaptic projections to sympathetic premotor neurons for regulating BAT thermogenesis 63 (Figure 3(A)). GLP-1 administers into DMH increased BAT thermogenesis, while GLP-1R knockdown in DMH increased adiposity, along with a reduction in energy expenditure, BAT temperature, and UCP-1 expression. 63 Therefore, GLP-1R signaling in DMH is well placed to regulate energy balance by controlling sympathetic outflow and BAT function.
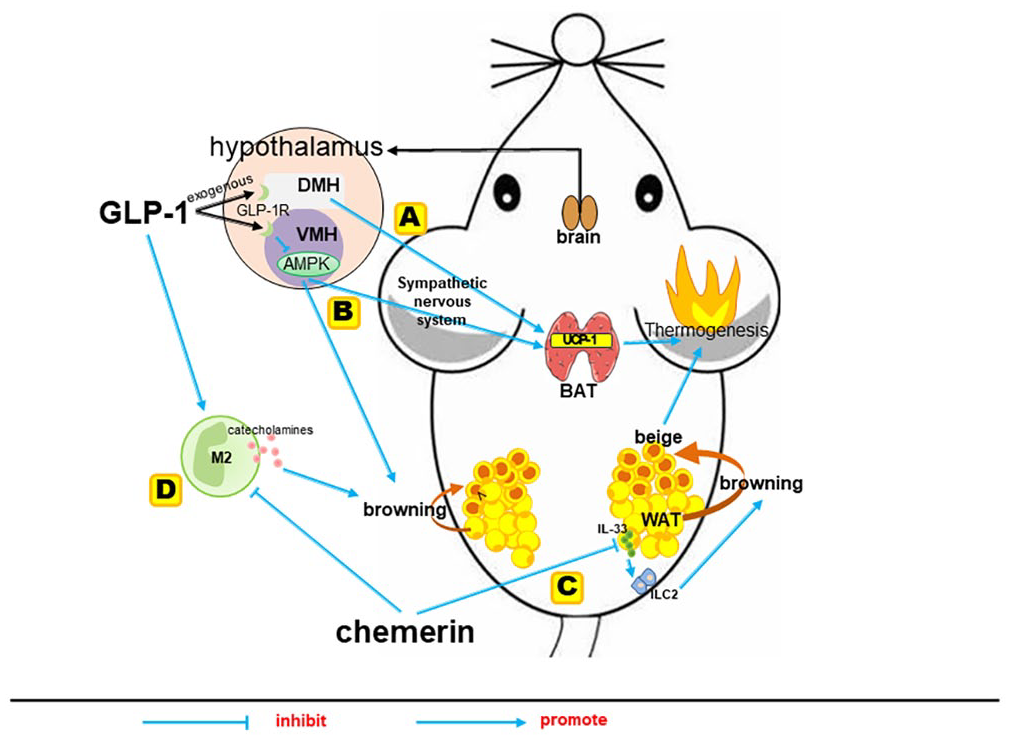
The regulation mechanisms of GLP-1 and chemerin on browning of WAT and thermogenic gene UCP-1 level. (A and B) GLP-1 was well placed to regulate energy balance by controlling sympathetic-induced BAT thermogenesis and browning of WAT. (C) Chemerin–CMKLR1 axis negatively regulated type 2 innate immune responses via targeting adipocytes to restrict IL-33 production, thereby limiting the browning of WAT and thermogenesis. (D) The M2 macrophages induced local production of catecholamines, which is the dominant trigger required for the conversion of WAT into beige adipocytes.
In addition to DMH, specific injection of liraglutide in the ventromedial hypothalamus (VMH) can trigger the inhibition of AMP-activated protein kinase (AMPK) and lead to sympathetic-induced BAT thermogenesis and browning of WAT. 62 AMPK is an important mediator controlling energy dissipation and a key negative regulator of sympathetically activated BAT thermogenesis, integrating peripheral signals such as thyroid hormone and leptin. 64 VMH neurons modulate the sympathetic nervous system in brainstem areas such as raphe pallidus and inferior olive, which have been functionally linked to the regulation of BAT thermogenesis. 64 Intracerebroventricular liraglutide administration decreased the AMPK activity in the hypothalamus, while the activation of AMPK in the VMH abolished the actions of liraglutide on BAT thermogenesis and WAT browning, indicating that brain GLP-1/GLP-1R actions on BAT thermogenesis and WAT browning are mediated by the inactivation of AMPK in the VMH of the hypothalamus 62 (Figure 3(B)).
Chemerin inhibits thermogenesis
When the lipid distribution of 3T3-L1 adipocytes changed from large droplets to a multilocular distribution, the mRNAs of brown/beige adipocytes UCP-1 were increased, while the mRNA of chemerin was reduced. 67 Moreover, contrary to WT mice treated by dihydrotestosterone (DHT), CMKLR1 KO mice treated by DHT had smaller adipocytes, decreased fatty acid–binding protein 4 (FABP4) expression level, increased mRNA expression of PGC-1α, irisin, and UCP-1. 68 It is known that FABP4, one of the adipokines, is regarded as a WAT marker. Whereas PGC-1α and irisin, both of which are regarded as the markers of beige cells, could stimulate the expression of UCP-1. So, these results indicated that epididymal white fat cells of CMKLR1 KO mice were more easily converted into beige fat cells, confirming the involvement of chemerin/CMKLR1 pathway in the browning of WAT. 68 A study recently identified that the adipose chemerin–CMKLR1 axis was a physiological negative regulator of thermogenic beige fat biogenesis. 51 Global or adipocyte-specific ablation of the chemerin–CMKLR1 axis promoted cold-induced WAT browning and adaptive thermogenesis, while it did not affect the expression of UCP-1 and other thermogenic genes in classical BAT upon cold exposure, suggesting that such inhibitory effects are specific to beige fat formation. 51 In addition to its impact on WAT browning, chemerin/CMKLR1 has also been reported to play a role in regulating BAT thermogenesis.69,70 For instance, both male and female chemerin KO mice exhibit a noticeable increase in BAT mass, accompanied by an augmentation in the size of brown adipocytes. 70
About the mechanism of chemerin affecting thermogenesis, it has been reported that the enhanced cold-induced thermogenic beige fat biogenesis caused by ablation of the chemerin–CMKLR1 axis was dependent on the elevated IL-33 production and subsequent activation of type 2 innate immune responses in WAT. IL-33, mainly produced by WAT, is a critical upstream cytokine to initiate type 2 immune responses primarily via activation of type 2 innate lymphoid cells (ILC2) and subsequent secretion of bioactive molecules.71,72 The lack of the chemerin–CMKLR1 axis in WAT promotes cold-induced IL-33 production and type 2 innate immunity and enhances cold-induced beige fat biogenesis and thermogenesis. In addition, the neutralization of IL-33 or depletion of ILC2 abrogates the enhancement of cold-induced beige fat biogenesis in chemerin KO mice. 51 Since WAT is relatively poorly innervated, recent studies pay more attention to the critical role of type 2 innate immunity in facilitating beige fat development. 73 Type 2 cytokine-associated innate immune cells, including ILC2, M2 macrophages and eosinophils, are enriched in the WAT of lean, but in obesity, type 2 cytokine-associated immune cells shift to type 1. 74 In view of this, Lin et al. 51 proposed a feed-forward circuit between adipocytes and type 2 innate immunity via IL-33, which is critical for thermogenic beige fat formation in vivo, and revealed that the chemerin–CMKLR1 axis negatively regulated such circuit via targeting adipocytes to restrict IL-33 production, thereby limiting the browning of WAT and thermogenesis (Figure 3(C)).
Not only ILC2 but also M2 macrophages, another type 2 cytokine-associated innate immune cells, were found to increase in chemerin KO mice and CMKLR1 KO mice. Interestingly, multiple studies have shown that M2 macrophages in adipose tissue play a significant role in the browning of WAT.75–77 Injecting M2 macrophages of adipose tissue into DIO adult WT mice effectively triggered their WAT browning, reduced their pro-inflammatory responses, and improved their insulin sensitivity. 75 A study in vivo showed that cold stimulation to obese mice activated M2 macrophages and promoted browning of WAT, effectively inducing the thermogenesis of adipose tissue and increasing energy expenditure. 76 Activated M2 macrophages induced tyrosine hydroxylase expression and local production of catecholamines. 77 Specifically, the release of norepinephrine is the dominant trigger of cold-induced beige fat development. 60 Beige adipocytes specifically increase the expression of UCP-1 in response to environmental cold and further increase energy expenditure throughout the body. 78 Considering the important roles of M2 macrophages in the WAT browning process and the modulations of GLP-1 and chemerin on the levels of M2 macrophages, we speculate that WAT browning and thermogenesis enhanced induced by GLP-1 and chemerin are likely to be mediated through promoting macrophage to M2 polarization (Figure 3(D)), but it needs further confirmation.
Interplay between GLP-1 and chemerin in vivo
Downregulation of chemerin expression by GLP-1
In 2015, the first report emerged indicating that GLP-1R agonist liraglutide could effectively reduce chemerin expression in the livers of rats. A subsequent four-week liraglutide treatment of HFD-induced insulin-resistant rats not only alleviated endoplasmic reticulum (ER) stress in the liver but also led to a simultaneous reduction in both liver and circulation-based chemerin expression, accompanied by a notable improvement in insulin sensitivity. 11 Importantly, ER stress was shown to promote the dissociation of glucose-regulated protein 78 (GRP78), a pivotal pathway implicated in the induction and progression of IR. Intriguingly, the mRNA expression of chemerin appeared to correlate significantly with the mRNA levels of GRP78, highlighting chemerin’s potential role as a key link between ER stress and IR. Yang et al. 11 postulated that GLP-1 may influence the level of chemerin by affecting ER stress and IR.
Furthermore, a study conducted on diabetic patients has suggested that the reduction of chemerin levels may not be directly regulated by GLP-1, but it rather linked to changes in body composition, such as weight loss and fat loss. 79 Interestingly, another study concluded that the weight-reducing effect of GLP-1 is unlikely to be the primary reason for the changes in chemerin observed in DIO mice. Notably, PEX-168, the first long-acting GLP-1R agonist independently developed in China and covered by the Chinese health insurance system, demonstrated significant effects in reducing chemerin levels, body weight, improving IR, reducing inflammatory reactions, and preventing the development of prediabetes. 4 Remarkably, despite the absence of weight loss in the DIO mice treated with a low dose of PEX-168 at the end of the intervention, the levels of blood glucose, IR, and chemerin were notably improved, and these improvements were consistent across different doses. 4 In this study, IR exhibited a highly significant correlation with chemerin, leading Guo et al. 4 to infer that the regulation of chemerin expression by PEX-168 may be closely associated with its hypoglycemic effect.
Another study has reported that the expression of chemerin may be directly reduced by GLP-1R agonists. For instance, a six-week intervention with a dipeptidyl peptidase-4 (DPP-4) inhibitors, which can preserve high circulating levels of intact GLP-1, was found to significantly decrease chemerin levels in rats with T2DM. Furthermore, it was observed that the effect of the DPP-4 inhibitor in reducing chemerin could be partially reversed by a phosphoinositide 3-kinase (PI3K) antagonist. 5 The PI3K–AKT signaling pathway is known to play a crucial role in mediating the biological signals of insulin in vivo, suggesting that DPP-4 inhibitors may regulate chemerin expression in visceral adipose tissue through this pathway. 5 In conclusion, GLP-1-based therapies, including GLP-1R agonists and DPP-4 inhibitors, hold promising potential in modulating chemerin expression, which may play a vital role in managing obesity and T2DM. Nevertheless, further investigation is warranted to precisely elucidate the mechanisms underlying the relationship between GLP-1 and chemerin, thus offering promising therapeutic avenues for managing obesity and related metabolic disorders.
Inhibition of GLP-1 synthesis and secretion by chemerin
Ernst et al. 80 found that exogenous chemerin reduced glucose uptake in diabetic mice and exacerbated glucose intolerance. In addition, long-term HFD-fed CMKLR1 KO mice exhibited additional metabolic benefits compared to controls, with significantly improved glucose tolerance. 51 These results imply a potential role of chemerin in regulating pancreatic β-cell function. Our recent study further confirmed the beneficial effects of adipose tissue–specific chemerin KO on pancreatic β-cell morphology and function in HFD-fed mice. Adipose chemerin deletion restored the damaged pancreatic islet structure induced by the HFD and significantly improved the function of the pancreas in HFD-fed mice (as evidenced by markedly decreased blood glucose levels at 30, 60, and 120 min after oral glucose administration compared to control mice, and a significantly lower area under the glucose tolerance curve). 12 Consequently, we then asked the potential mechanisms through which chemerin modulates pancreatic β-cell morphology and function. GLP-1 is a peptide hormone secreted by intestinal L-cells in response to postprandial glucose levels, stimulating insulin secretion in a glucose-dependent manner and promoting β-cell proliferation to improve its blood glucose-lowering function. Interestingly, chemerin expression has been identified in the colon 81 and plays a significant role in regulating intestinal inflammation and gut microbiota. 82 Given this, we hypothesize that chemerin might also affect GLP-1 secretion in the intestine.
Recently, our research has demonstrated that aerobic exercise significantly increases the mRNA and protein levels of GLP-1 in the colon of diabetic mice, leading to enhancements in pancreatic β-cell morphology and glucose regulation. Furthermore, both serum and colon chemerin levels were markedly reduced. However, exogenous chemerin supplementation weakens the beneficial effects of exercise on GLP-1 levels and pancreatic β-cell morphology and function, indicating a potential association between the changes in colon GLP-1 levels and the role of chemerin reduction in the improvement of pancreatic β-cell structure and function in aerobic exercise–treated diabetic mice. 12 Furthermore, we assessed the GLP-1 levels in heterozygous and homozygous adipose chemerin KO mice and observed significant increases in colon and serum GLP-1 levels in homozygous mice, providing the first in vivo evidence of chemerin’s negative regulatory influence on GLP-1. Given the significant roles of chemerin and GLP-1 in regulating pancreatic morphology and function, along with chemerin’s negative regulatory impact on GLP-1 levels, it indicates that likely exerts its effects on improving pancreatic structure and function by elevating colon and serum GLP-1 levels. 12 In the future, combining histological examination of colon tissues with the analysis of relevant inflammatory factors may further elucidate the specific mechanisms through which chemerin regulates GLP-1 secretion.
Conclusions
This review summarized the contrary effects and their mechanisms of GLP-1 and chemerin on obesity development, including known understanding (such as regulating insulin secretion and insulin sensitivity, glycolipid metabolism, and appetite) and some new insights (GLP-1 increasing anti-inflammatory M2 macrophages in adipose tissue and promoting WAT browning and thermogenesis, while chemerin acting the opposite way). Besides, the relationship between GLP-1 and chemerin was summarized briefly. Considering the multifaceted beneficial impact of GLP-1 on adipose tissue in obesity, its regulation of the adipokine chemerin opens up novel research avenues to explore the underlying mechanisms. Furthermore, as an inflammatory factor capable of acting within colonic tissue, chemerin offers a new pathway for investigating the influencing factors on colonic GLP-1 secretion during obesity. This is beneficial to deepen the understanding of endocrine mechanisms of obesity and to provide strategies for the treatment of obesity and related diseases.
Footnotes
Authors’ Contributions
All authors searched and analyzed retrieved literature, prepared figures, drafted sections of the text, and revised the draft manuscript for critical intellectual content. All authors jointly decided to submit the final manuscript for publication.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (grant no. 31872801) and the Shanghai Frontiers Science Research Base of Exercise and Metabolic Health, Shanghai University of Sport.