
Abstract
Amyloidosis comprises a spectrum of protein misfolding disorders characterized by the aggregation of diverse amyloidogenic proteins into cytotoxic fibrillar structures. Due to the conserved β-sheet-rich architecture of fibrils across various amyloid species, pan-amyloid therapeutics present a promising strategy for simultaneously targeting multiple amyloidogenic proteins. In this study, four representative amyloidogenic proteins—amyloid β, serum amyloid A1, islet amyloid polypeptide, and transthyretin—were selected as targets. From a library of 10,272 small molecules, five functional groups were identified as critical contributors to high-affinity binding. Utilizing these functional groups, potential compounds capable of binding to all four amyloidogenic proteins were further screened. This functional group-based screening workflow not only validates the effectiveness of functional group-driven screening in multitarget drug discovery but also facilitates the efficient identification of candidate pan-amyloid binding compounds. Furthermore, these findings provide valuable insights into the development of pan-amyloid therapeutic strategies.
INTRODUCTION
Amyloidosis encompasses a group of protein-misfolding disorders characterized by the aggregation of >40 distinct proteins into cytotoxic forms within vital organs.1–4 The phenotypes of these diseases vary significantly, manifesting as neurodegenerative conditions such as Alzheimer’s disease (amyloid-β [Aβ], tau)5,6 and Parkinson’s disease (α-synuclein), 7 as well as systemic pathologies like transthyretin amyloidosis (ATTR) 8 and metabolic dysregulation associated with type 2 diabetes (amylin). 9 Despite their diversity, all these conditions share a common aggregation pathway: soluble monomers undergo conformational changes to form β sheet-rich intermediates, which then assemble into mature amyloid fibrils through conserved nucleation, elongation, and secondary nucleation mechanisms.10,11 This structural convergence presents a unique therapeutic opportunity to target all forms of amyloid proteins, regardless of the specific misfolded protein species, a concept known as pan-amyloid therapeutics. 12 Furthermore, emerging evidence suggests that small molecules targeting universal fibril architectures (e.g., parallel β-sheet grooves and salt-bridge networks), rather than sequence-specific epitopes, may provide broader therapeutic effects across various amyloid disorders. 13
Current therapeutic paradigms continue to emphasize single-target approaches, exemplified by monoclonal antibodies targeting Aβ (such as aducanumab) and transthyretin stabilizers (like tafamidis). While these agents demonstrate modest clinical benefits, they have limitations in addressing several challenges. First, cross-seeding phenomena among amyloid species (e.g., Aβ/hIAPP, Aβ/tau, Aβ/α-synuclein) indicate molecular cross-talk between distinct amyloid diseases, suggesting that targeting only one species may be insufficient. 14 Second, the amyloid aggregation network involves compensatory or alternative aggregation pathways, with a single protein species often functioning as a seed, product, intermediate, or catalyst. This complexity highlights the limitations of targeting individual reaction nodes. 15 Third, in aging populations, multisystem amyloid deposition affecting organs such as the heart, kidneys, liver, nervous system, and digestive tract is common, particularly in wild-type ATTR and other systemic amyloidosis. 16 These challenges underscore the necessity for pan-amyloid strategies. Consequently, there is growing interest in identifying pan-amyloid inhibitors capable of broadly disrupting amyloid aggregation across multiple pathogenic proteins to address the complexity of amyloid-associated pathologies. Currently, several small aromatic molecules, including polyphenols, 17 have been identified as effective inhibitors of the aggregation of various amyloid peptides. 18
The binding of a molecule to its target is widely recognized as the initial and rate-limiting step that determines its biological effects. 19 Therefore, it is essential for aggregation inhibitors to bind to amyloidogenic proteins. 20 Based on this premise, we developed a functional group-centric virtual screening framework designed to identify candidate small molecules with broad-spectrum anti-amyloid activity. Unlike traditional ligand-based or single-target approaches, this strategy focuses on conserved structural motifs across amyloidogenic proteins, thereby increasing the likelihood of discovering universal inhibitors. The approach comprises three key components: (1) the construction of an initial compound library (CL-I) containing 10,272 small molecules retrieved from the PubChem database using the keyword “amyloid aggregation inhibition.” (2) Multitarget molecular docking was performed against four representative amyloidogenic proteins—Aβ, serum amyloid A1 (SAA), islet amyloid polypeptide (IAPP), and ATTR—followed by binding interaction analysis to identify functional groups critical for high-affinity binding; and (3) the generation of a refined compound library (CL-II) enriched with these functional groups. Using the constructed datasets, we then conducted comparative docking analyses and hierarchical subset selection to identify promising candidates with potential pan-amyloid inhibitory activity.
This strategy ultimately identified five functional groups—carboxamide, tetrahydroacridine, imidazole, trifluoromethoxy, and indole—that were highly enriched in potent binders. Subsequent analysis of their prevalence and distribution revealed that two of these groups, indole and imidazole, exhibited consistent structural convergence across all high-affinity compounds, underscoring their central role as pharmacophores for pan-amyloid inhibition. By validating this approach through statistical analysis of binding energy shifts and hierarchical subset selection, we not only demonstrated the feasibility of functional group-based virtual screening but also established a scalable, target-agnostic framework for future drug discovery efforts targeting amyloid-related diseases. This finding also lays the foundation for the rational design of broad-spectrum therapeutics using highly efficient functional group-driven virtual screening strategies.
MATERIALS AND METHODS
Selection of Screening Amyloidogenic Proteins
In this study, we aim to identify compounds that can effectively inhibit the aggregation of amyloidogenic proteins. To comprehensively evaluate the potential inhibitory capabilities of these compounds, we have selected four common amyloidogenic proteins as targets for virtual screening, including Aβ, SAA, IAPP, and ATTR. These proteins are characterized by a high content of β-pleated sheets in their structures and are associated with various diseases. As illustrated in Supplementary Table S1, the selected target structures encompass various aggregation states of amyloids. This structural diversity among the targets enhances the validity of the virtual screening results for identifying pan-amyloid inhibitors.
Four PDB structure files of the selected amyloidogenic proteins were downloaded from the RCSB Protein Data Bank. Water molecules and other ligands were removed from the PDB files, and polar hydrogen atoms were added to the protein structures. Finally, the structures were saved in PDBQT format, which can be represented as a kinematic tree of the molecular receptor file using AutoDockTools. 21 The resulting structures were utilized for subsequent molecular docking.
Construction of Compound Libraries
In this study, we constructed two distinct databases to effectively identify pan-amyloid inhibitors and validate a functional group-driven search strategy. To identify function groups, we created an initial compound library (CL-I) by querying the PubChem database with specific keywords. A total of 10,272 small molecules were identified, and their structures, along with relevant information such as molecular names and inhibitory activities, were extracted from the database using web crawling technology. The 3D structure of each molecule was generated using OpenBabel software. 22
To efficiently and accurately identify potential pan-amyloid inhibitors, we further constructed a refined compound library II (CL-II) based on five functional groups derived from the initial screening of CL-I. Specifically, we conducted a search in the PubChem database using a combinatorial query that incorporated these five key functional groups. We established the selection criteria for CL-II as molecules containing at least three of the five identified functional groups. By employing substructure search algorithms and SMARTS pattern matching, we retrieved 5,623 compounds that met this condition. The resulting library was formatted in PDBQT to ensure compatibility with AutoDock Vina docking protocols. Figure 1 presents a comprehensive overview of the screening process.
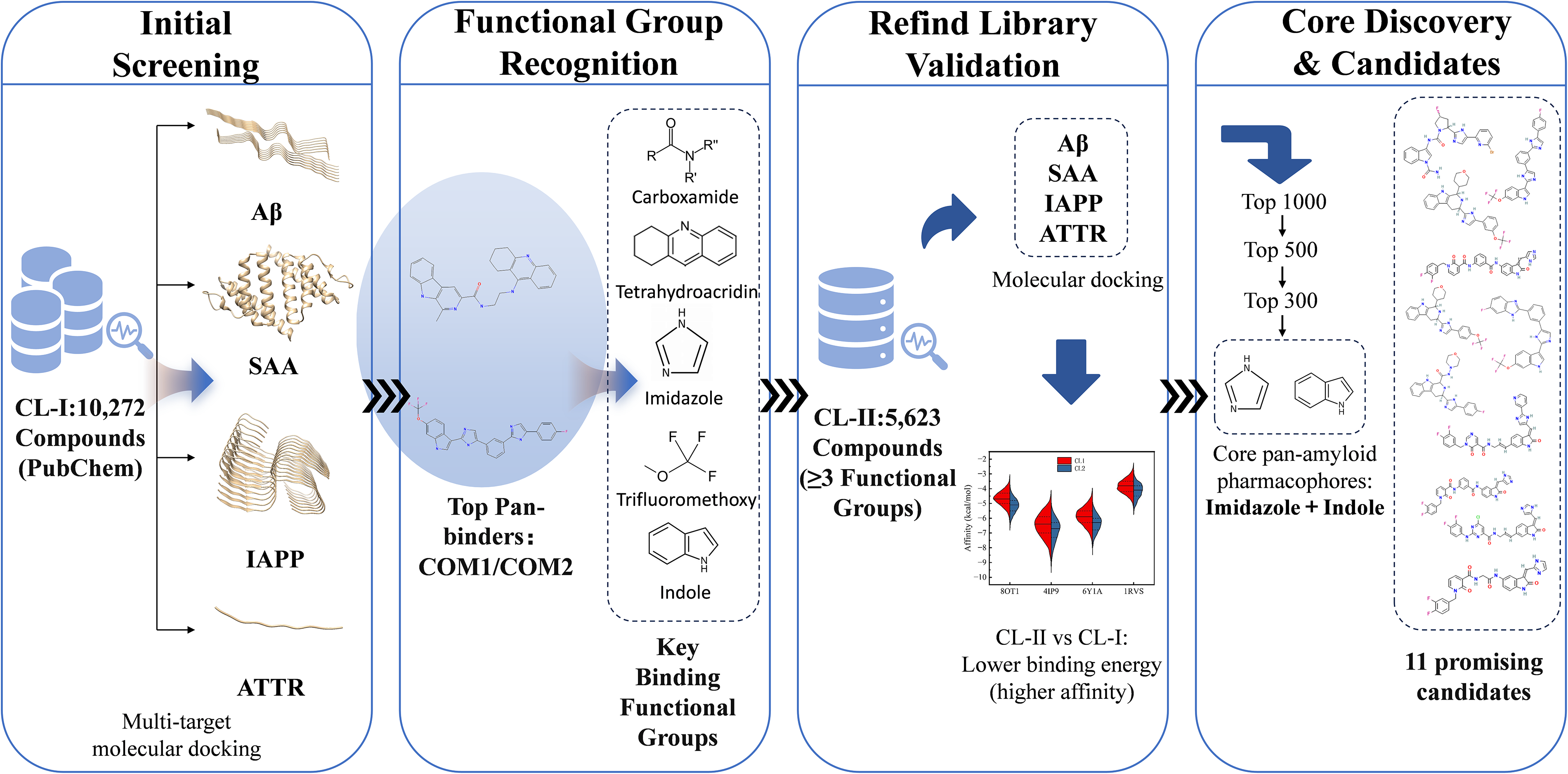
The complete workflow for functional group-based virtual screening.
Molecular Docking
Over the past few decades, numerous docking software packages have been developed that employ different scoring functions or search algorithms, including AutoDock4, 23 AutoDock Vina (Vina), 24 PSO@AutoDock, 25 MetaDock, 26 and FIPSDock. 27 Among these methods, Autodock Vina exhibits exceptional docking performance, owing to its search algorithm, which effectively integrates the global Markov Chain Monte Carlo method with local Broyden–Fletcher–Goldfarb–Shanno optimization. Currently, AutoDock Vina has become a standard virtual screening tool in contemporary drug discovery due to its speed, accuracy, and user-friendliness, making it particularly well-suited for high-throughput screening. Consequently, in this study, we utilized AutoDock Vina as docking tool to screen compounds that bind to various amyloidogenic proteins.
Prior to conducting molecular docking, it is essential to identify docking positions for the targeting amyloidogenic proteins. We determined the docking positions based on several guiding principles. First, we analyzed four protein structures using the CavityPlus web server 28 to identify potential docking sites. A cavity in a protein is classified as having “medium” druggability if it is recognized as a suitable docking position, as demonstrated in the cases of 8OT1 and 4IP9. Second, for the protein structure 6Y1A, CavityPlus did not indicate the presence of cavities suitable for drug targeting. It has been established that the formation of β-sheet structures is crucial for the aggregation of amyloidogenic proteins.29,30Furthermore, amyloid fibrils are formed through the binding of monomers to their surfaces: molecular dynamics (MD) and kinetic studies have demonstrated that monomer-fibril surface interaction drives both fibril elongation and secondary nucleation, meaning that the fibril surface actively promotes additional aggregate formation.31,32 Consequently, we identified a portion of the fibril surface composed of β-strands as potential docking sites. Last, for 1RVS, which has a relatively simple structure, we analyzed the characteristics of its constituent amino acids. Aromatic amino acids play a crucial role in amyloid aggregation due to their unique π–π interactions and hydrophobic properties. Tyrosine, in particular, features a hydroxyl group adjacent to its aromatic ring, enabling it to participate in π–π stacking while potentially enhancing stability through hydrogen bonding. Therefore, we designated the region surrounding the tyrosine side chain of the amyloid as the docking site. According to the principles outlined above, a box measuring 60 × 60 × 60 Å 3 was placed in the designated regions for the four proteins. The detailed composition of the docking sites for these proteins is illustrated in Supplementary Table S2.
Each compound was randomly assigned to the designated docking site of amyloidogenic protein structures. By continuously altering the binding conformations, the binding affinities for each docked compound were evaluated. In this study, the top 10 binding conformations were retained for each compound. After the docking process was completed, the conformation with the lowest binding energy was selected as the optimized conformation for further analysis.
MD Simulation
We performed MD simulations to calculate the binding energy between the protein and ligand. The bound protein–ligand complex obtained from docking was used as the initial structure for these simulations. This structure was placed in a cubic box with a minimum distance of 1.0 nm from the box edges. The box was then filled with TIP3P water molecules and counter ions to neutralize the system’s charge. Prior to the MD run, energy minimization was conducted to relieve any structural clashes, followed by equilibration runs under NVT conditions at 300 K and NPT conditions at 1 bar. Finally, the MD simulation was run for 50 ns under periodic boundary conditions. Binding energies between the protein and ligand were calculated using the Molecular Mechanics Poisson-Boltzmann Surface Area (MM-PBSA) method. 33 All MD simulations and analyses were performed using the GROMACS software package (version 5.0.1). The GROMOS96 43A1 force field was employed, and topology parameters for the ligand were generated using the ACPYPE server. 34
RESULTS AND DISCUSSION
Docking Results of CL-I with Amyloidogenic Proteins
The binding affinity vales obtained from molecular docking using AutoDock Vina directly reflect the interactions between small molecules and the target protein. This metric is considered the most critical factor in analyzing docking results and serves as a key criterion for evaluating the binding capacity of small molecules to proteins. Based on the premise that compounds inhibit amyloidogenic protein aggregation by initially binding to amyloidogenic proteins, we can specifically assess the binding affinity of small molecules to these proteins to preliminarily evaluate their potential to prevent aggregation.
In this study, an initial dataset (CL-I) comprising 10,272 small-molecule compounds was docked into four amyloid proteins to comprehensively evaluate the affinity of these small molecules for amyloids and to identify those that exhibit strong binding as potential aggregation inhibitors. The docking results reported multiple conformations along with their associated binding energies, from which the conformation with the lowest energy was selected. Table 1 presents information regarding the distribution of binding energies for the four proteins.
The Binding Energy of CL-I with Four Amyloidogenic Proteins
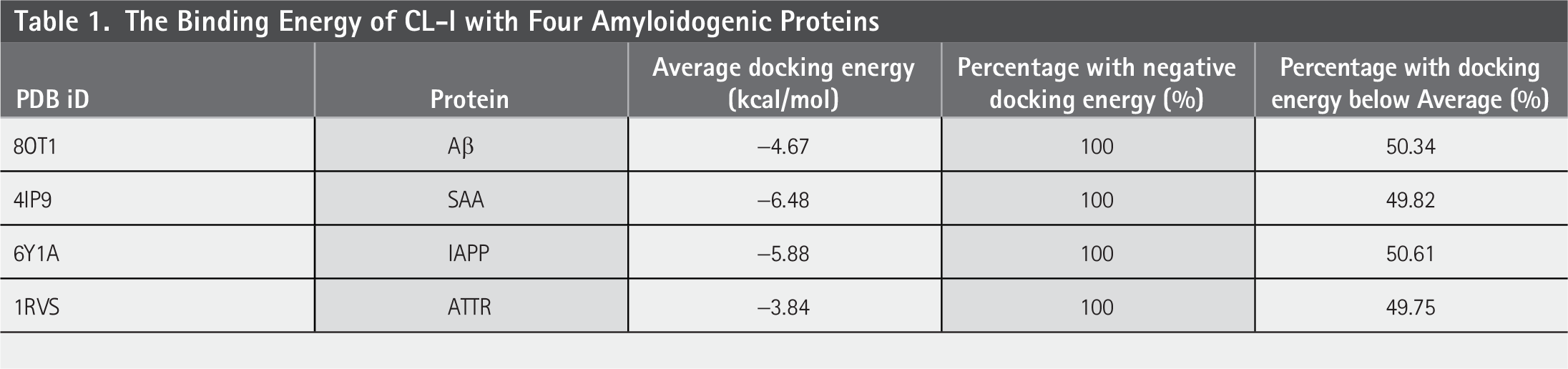
Table 1 shows that the binding energies of all molecules with the four amyloid proteins are negative, indicating that all candidates can bind, to some extent, to amyloidogenic proteins. Figure 2 illustrates the distribution of binding energies for 10,272 molecules across all amyloid protein structures. Overall, the shapes of distribution curves for the four proteins are similar and partially overlap. The docking energy distribution for 8OT1 and 1RVS are alike, exhibiting significantly narrower density curves. This indicates minimal differences in binding energy between these two target proteins and the small molecules. By comparing the central values of the energy distributions, the binding energy distribution center for 4IP9 falls within a more negative energy range, whereas the center for 1RVS corresponds to less negative values.
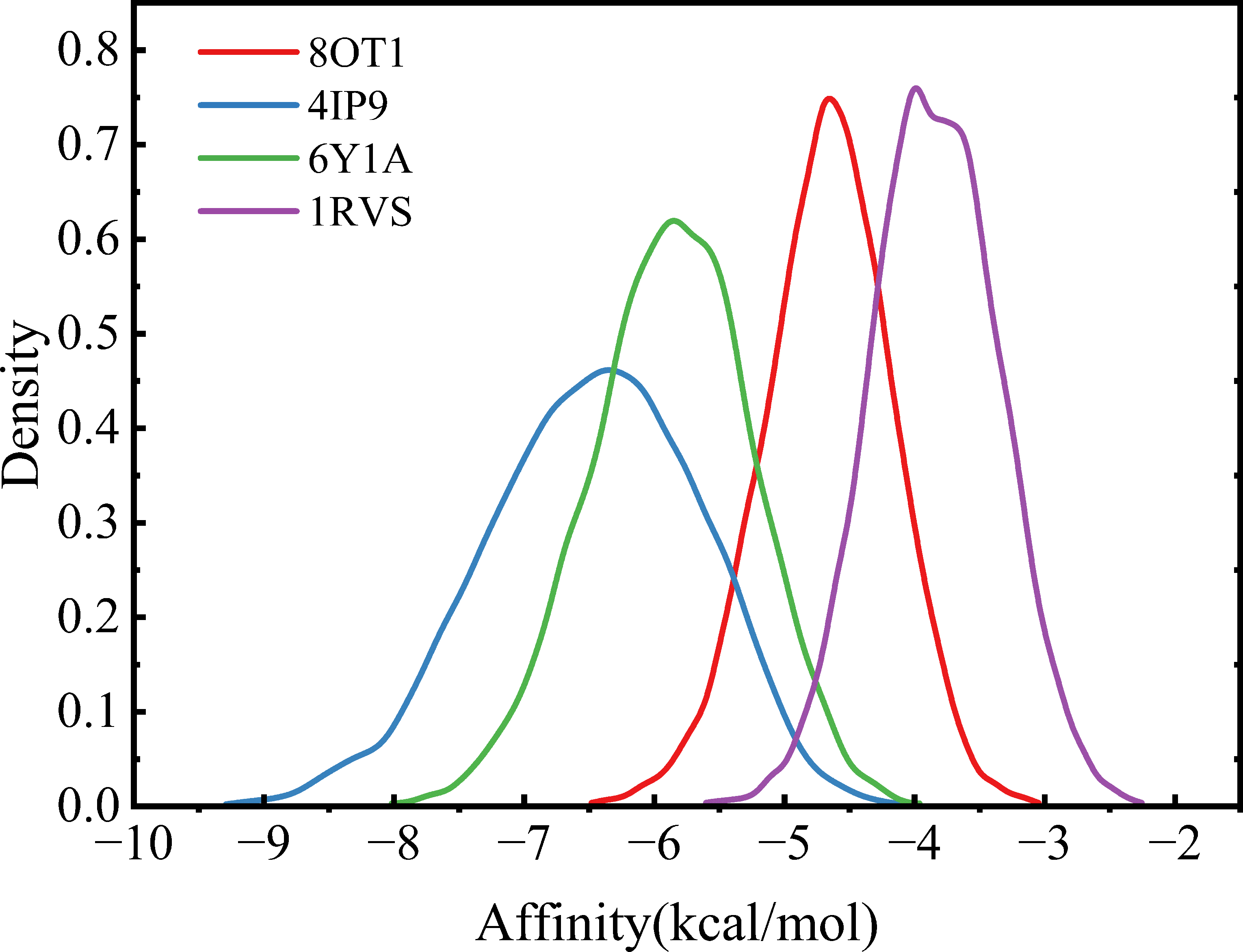
Density curves illustrating binding affinity of CL-I to four amyloidogenic proteins.
Identification of Functional Groups
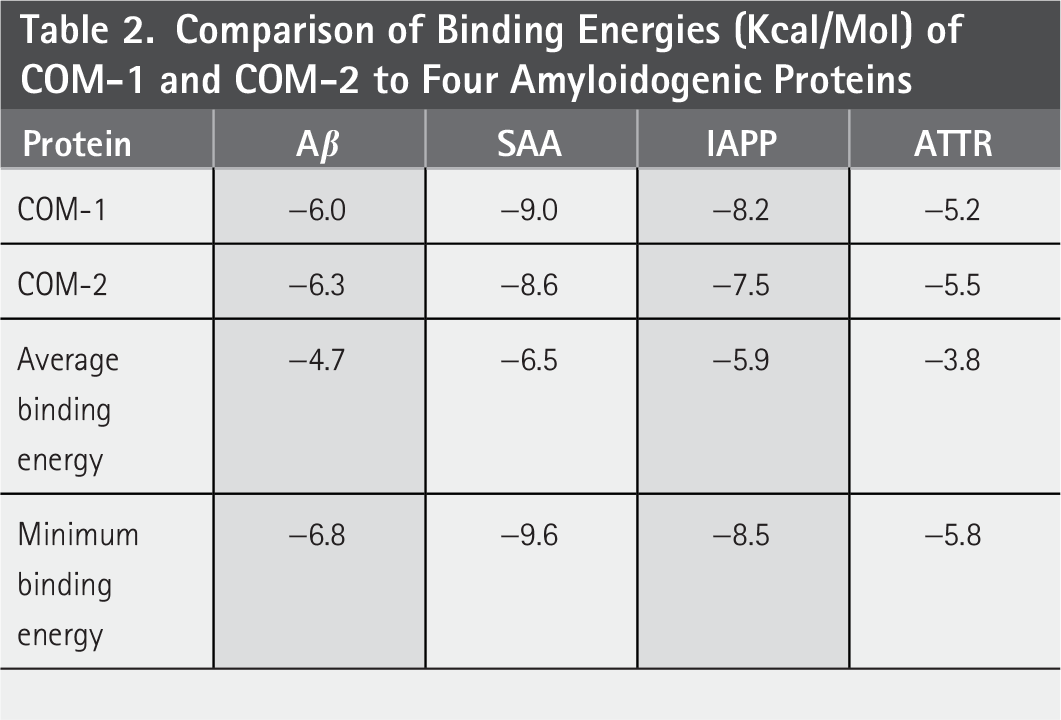
The 100 compounds with the lowest binding energies were considered potential inhibitors based on their affinity for specific amyloids. We compared these 100 compounds across four different amyloid types and identified two compounds that appeared in the rankings for all: COM-1 (SID312363223) and COM-2 (SID381870935). The docking energy results are summarized in Table 2. Both compounds exhibited binding energies below −5.0 kcal/mol for all four amyloid proteins, which is lower than the average binding energy and close to the minimum observed binding energy. These results indicate that two compounds have a strong affinity for the four target proteins and the potential to bind them, thereby inhibiting their aggregation and exerting the corresponding pharmacological effects.
By comparing the number of hydrogen bonds formed by the individual functional groups of COM-1 with each amyloid protein shown in Figure 3, we found that the nitrogen atom of the carboxamide group in the small molecule formed hydrogen bonds with three of the four amyloidogenic proteins (8OT1, 4IP9 and 1RVS). Additionally, the nitrogen atom of the tetrahydroacridine group formed hydrogen bonds with two of the four amyloidogenic proteins (8OT1 and 1RVS). These results suggest that these two chemical groups play a significant role in binding amyloidogenic proteins. Furthermore, the pyrido, ylamino, and indole groups form hydrogen bonds with Lys16 of 8OT1, Glu56 of 4IP9, and Asn64 of 6Y1A, respectively. Supplementary Figure S1 illustrates the detailed interactions between COM-1 and the four amyloidogenic proteins. Most importantly, the aromatic groups in COM-1 establish extensive hydrophobic interactions with hydrophobic residues within the binding cavity, which are crucial for amyloid binding. Based on the analysis above, incorporating carboxamid or tetrahydroacridine moieties into the small molecule is expected to enhance its binding affinity for amyloidogenic proteins.
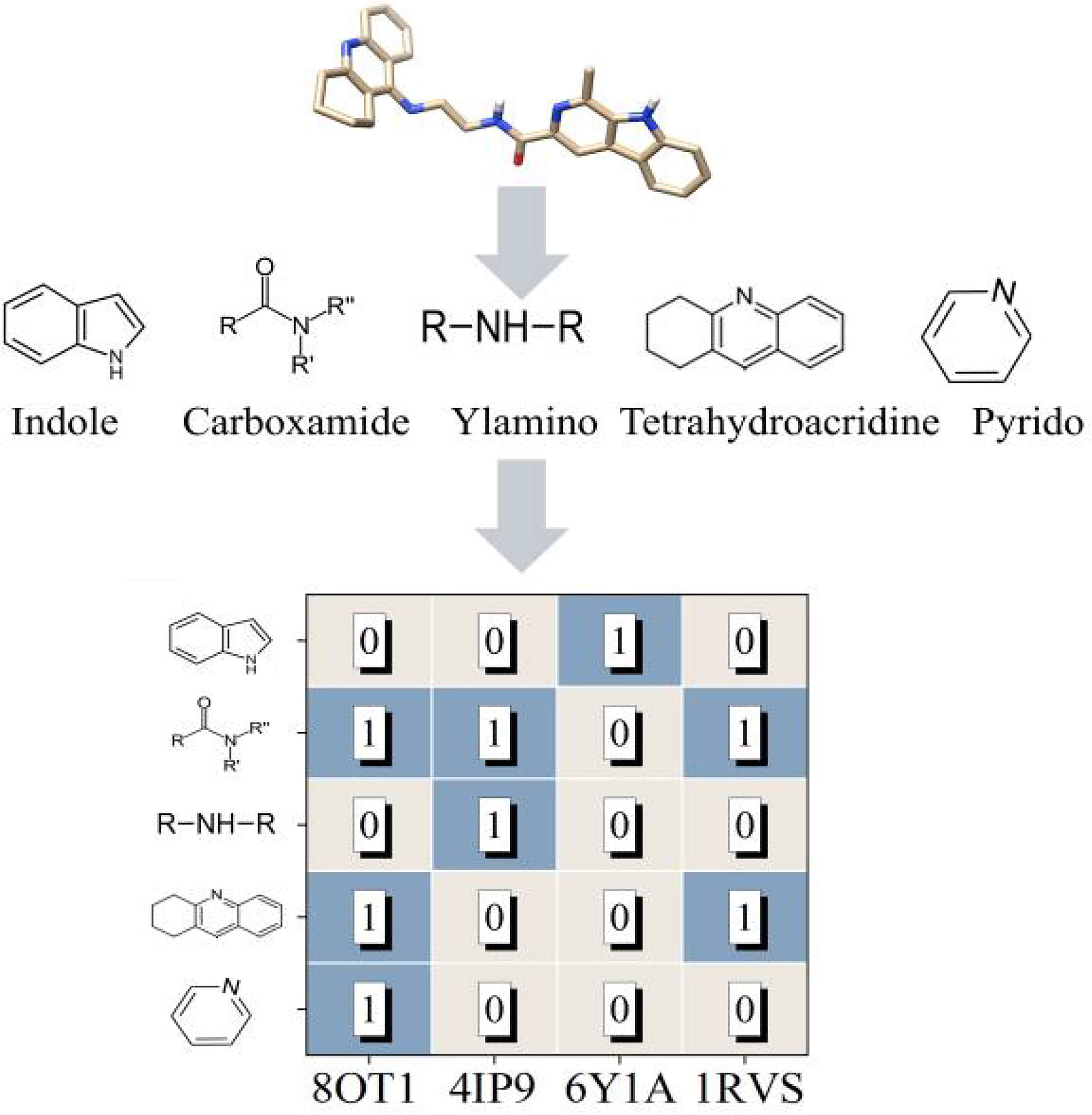
Functional groups present in COM-1 and the number of hydrogen bonds formed upon binding with four amyloidogenic proteins.
Similarly, COM-2 also contains five cyclic groups, and the presence of multiple rings may enhance various intermolecular interactions between the molecule and amyloid proteins. Among the binding conformations of COM-2 with the four amyloidogenic proteins shown in Figure 4, we observed that the nitrogen atom of the imidazole group forms hydrogen bonds with all four amyloidogenic proteins. The fluorine and oxygen atoms of the trifluoromethoxy group in the small molecule established hydrogen bonds with three of the four amyloids (8OT1, 4IP9, and 6Y1A). Notably, the same fluorine atom in the trifluoromethoxy group formed hydrogen bonds with both Ile26 and Asn22 of 6Y1A. Additionally, the nitrogen atom in the indole ring formed hydrogen bonds with two of the four amyloid proteins (4IP9 and 6Y1A). Supplementary Figure S1 also illustrates the detailed interactions between COM-2 and the four amyloidogenic proteins.
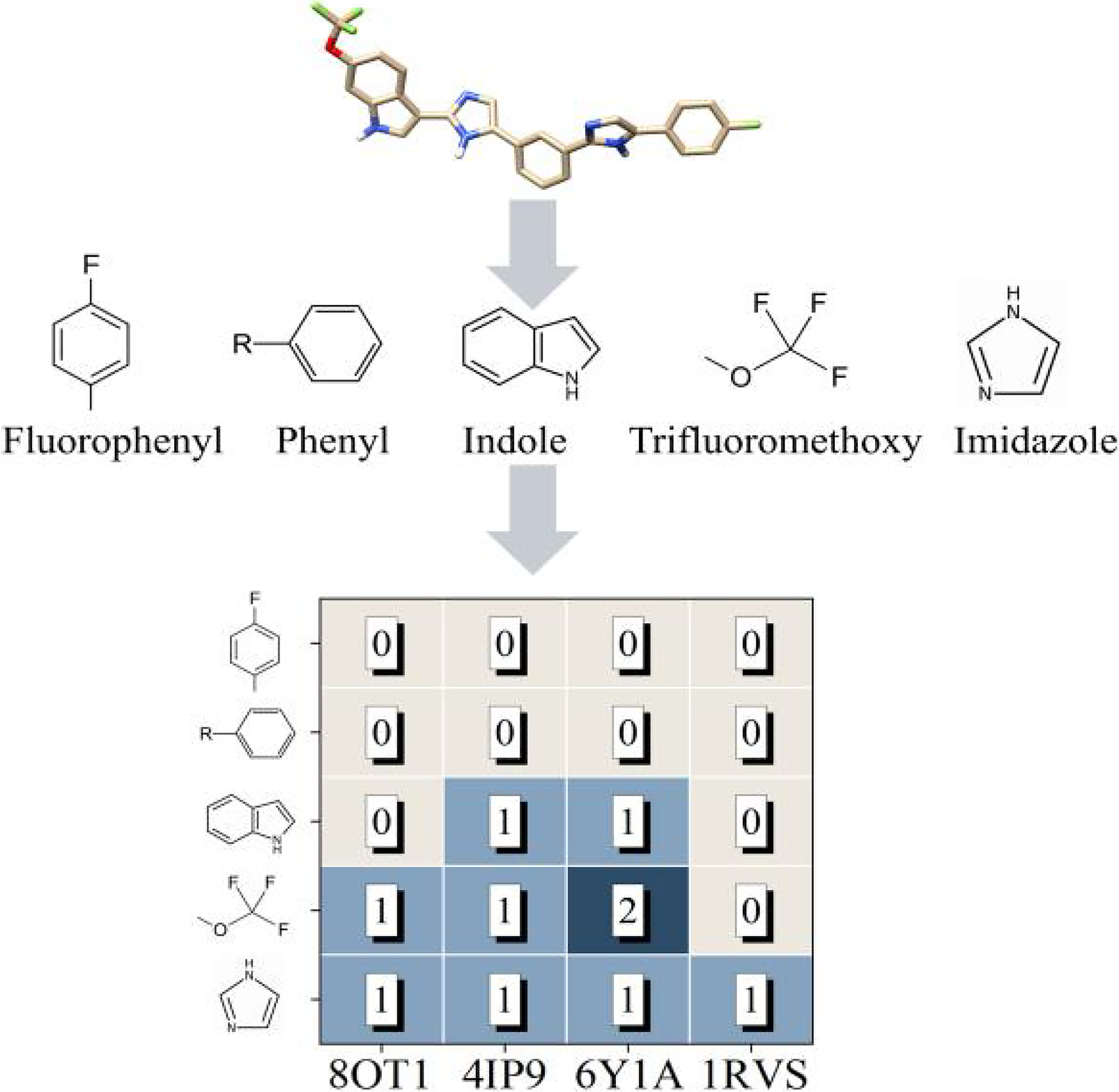
Functional groups present in COM-2 and the number of hydrogen bonds formed upon binding with four amyloidogenic proteins.
Based on the analysis of functional groups in COM-1 and COM-2, we concluded that five functional groups: carboxamide, tetrahydroacridine, imidazole, trifluoromethoxy, and indole, may play significant roles in binding to amyloid. It is anticipated that small molecules containing these five functional groups will exhibit above-average binding affinities.
Verification of Functional Groups
To verify the above hypothesis, we compiled a refined dataset comprising 5,623 compounds, each containing either three or four of the five identified functional groups. Supplementary Table S3 presents the distribution of these functional groups within the refined dataset. Using this dataset, we conducted docking analyses and compared the results with those obtained from the initial dataset shown in Figure 5. Compared to the initial dataset, the average docking energies of small molecules in the refined dataset were lower by 0.48 kcal/mol (8OT1), 0.36 kcal/mol (4IP9), 0.49 kcal/mol (6Y1A), and 0.31 kcal/mol (1RVS), respectively. The comparison of docking energies between the two datasets revealed that the functionally screened compound library (CL-II) exhibited superior binding affinity compared to the original library (CL-I) with mean binding energies of −5.63 kcal/mol and −5.22 kcal/mol, respectively. Therefore, the functional group-based screening strategy demonstrates enhanced molecular binding efficacy.
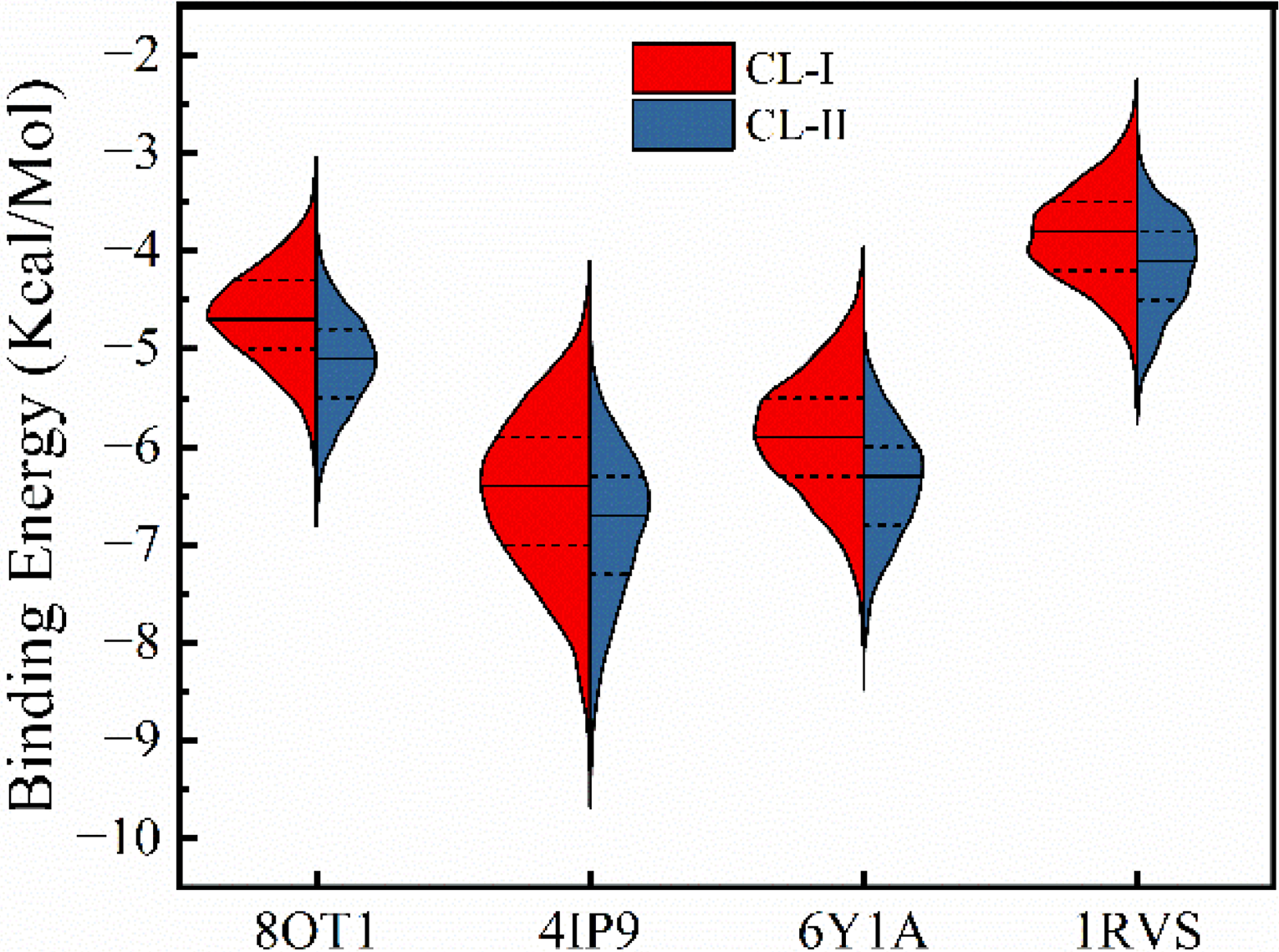
Comparison of the docking energies between CL-I and CL-II to four amyloidogenic proteins.
We further compared the distribution of docking energies between the two datasets. Statistical analysis revealed distinct distribution patterns, as summarized in Table 3. CL-I exhibited an approximately symmetric distribution (skewness = −0.1282) with slight left skewness and mild leptokurtosis (kurtosis = 0.1865), indicating a moderate prevalence of low-energy (high-affinity) binders and sporadic outliers. In contrast, CL-II showed significantly stronger left skewness (skewness = −0.2797) and platykurtic tails (kurtosis = −0.1488), suggesting a systematic enrichment of compounds with lower binding energies (i.e., higher predicted affinities) and reduced variability.
Statistical Analysis of Biding Energy Distribution of CL-I and CL-II to Four Amyloidogenic Proteins
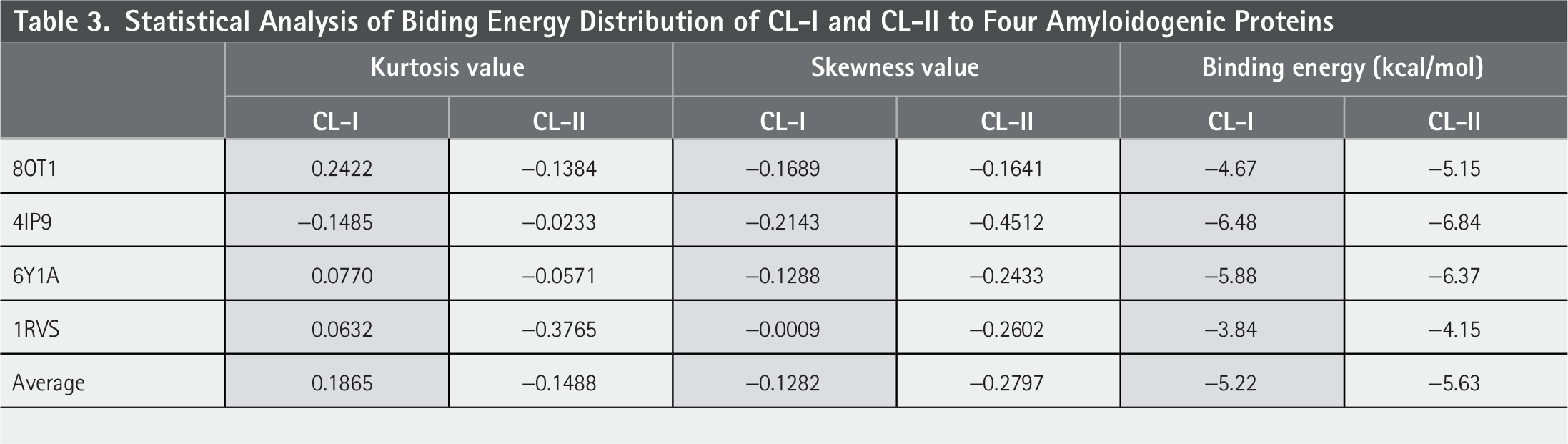
The results above demonstrate that the functional group-based strategy effectively prioritizes molecules with enhanced amyloid-binding potential. The observed shift toward lower binding energies in CL-II supports the hypothesis that targeted functional group selection improves inhibitory efficacy, likely by optimizing key interactions (e.g., hydrogen bonding, hydrophobic contacts) critical for amyloid aggregation. Further validation through in vitro assays is necessary to correlate these computational findings with biological activity.
Identification of Pan-Amyloid Inhibitors
The refined dataset exhibits relatively lower binding energies, indicating that the compounds included have the higher binding affinity to four amyloidogenic proteins. Based on the docking energies, three hierarchical subsets were generated by selecting molecules ranked in the top 1,000, 500, and 300 binding energies across all four targets, respectively.
The prevalence analysis of five functional groups was conducted for CL-II and three additional subsets. Prevalence was calculated as the percentage of compounds within each subset that contained a specific functional group. For each subset, the number of compounds featuring each of the five functional groups was counted and divided by the total number of compounds in that subset. This analysis highlights which functional groups become increasingly dominant as binding affinity thresholds become more stringent, providing insights into the structural features most critical for pan-amyloid inhibition.
Figure 6 illustrates significant evolutionary trends across five functional groups. The indole group demonstrated evolutionary dominance throughout the screening tiers, achieving 100% prevalence in the most stringent subset of the top 300 compounds (n = 13), suggesting its indispensable role in multi-target engagement. In contrast, the tetrahydroacridine group showed no enrichment, implying that its exclusion enhances pan-amyloid efficacy. Although the carboxamide group was nearly saturated in CL-II (99.1%), its prevalence declined to 69.2% in the top 300 subset, indicating that it functions as a permissive scaffold rather than a specificity driver. Its ubiquity in CL-II alone is insufficient to maintain affinity under competitive multitarget selection. The enrichment of imidazole (increasing from 71.9% to 84.6%) parallels that of indole, suggesting synergistic cooperation. The variable prevalence of trifluoromethoxy (47.6% in CL-II vs. 46.2% in the subset of top 500) indicates a context-dependent role, where its contribution to pan-amyloid inhibition may depend on specific structural partnerships (e.g., coexistence with the indole group) or target-specific binding pockets. These data identify indole as a central pharmacophore and imidazole as a cooperative partner in pan-amyloid inhibition.
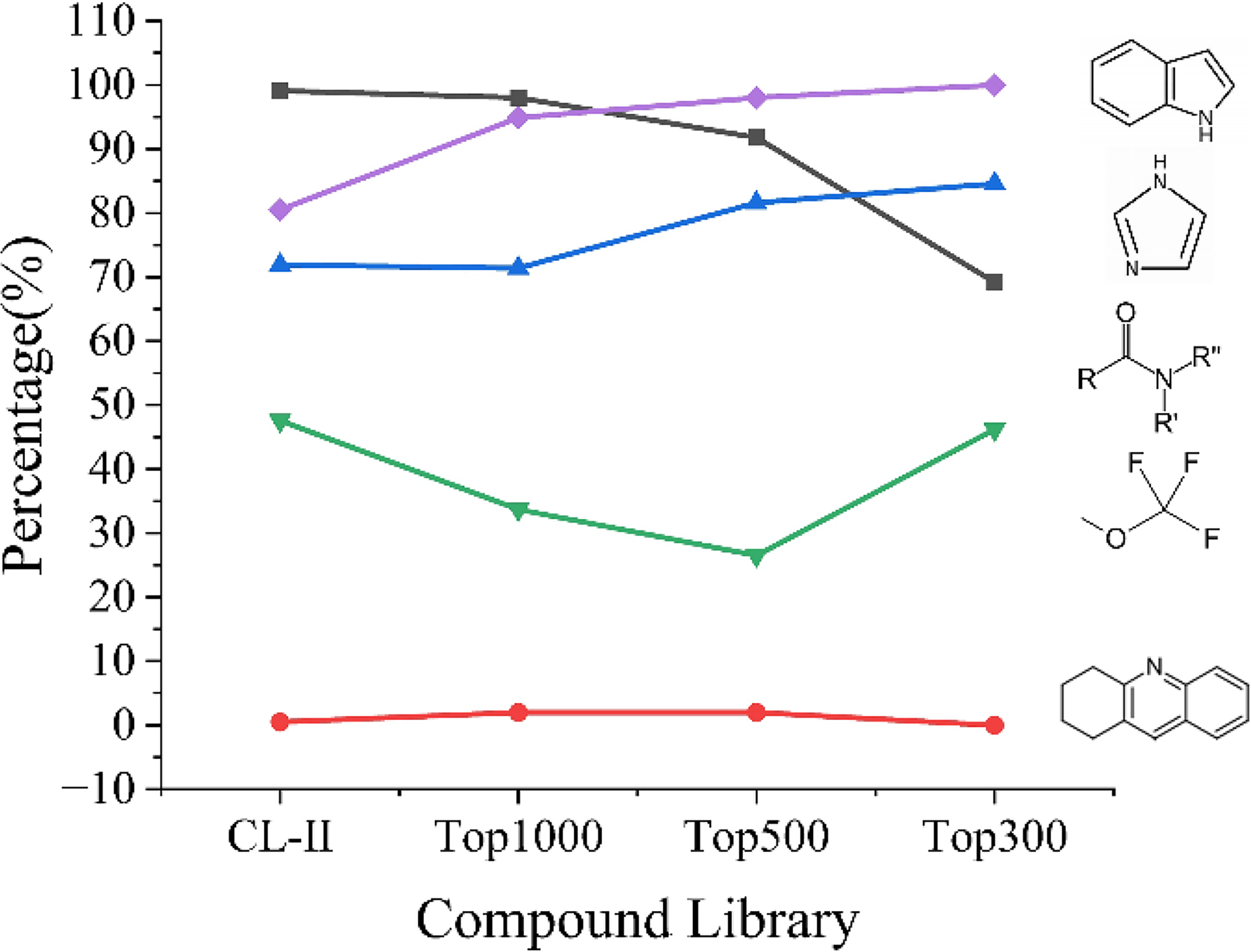
Evolution of the prevalence of five functional groups within CL-II and its three subsets.
Our conclusions are consistent with the findings of Prabir Kumar Gharai et al., who designed the amyloid inhibitor CT-01, 35 also incorporates imidazole and indole functional groups. This structural convergence underscores the critical importance of these two pharmacophores in pan-amyloid inhibition. Notably, CT-01 demonstrated broad-spectrum efficacy across multiple amyloid species in both in silico and in vitro assays, further validating the multitarget potential of imidazole and indole.
Based on the analysis above, we propose that small molecules containing both indole and imidazole moieties may exhibit the lowest binding energies and the greatest potential for broad-spectrum inhibition of amyloidogenic proteins. Accordingly, we identified 11 compounds within the top 300 subset that contain both indole and imidazole functionalities. Their molecular structures are shown in Figure 7, and their chemical nomenclature, along with docking energies for four amyloidogenic proteins, is listed in Supplementary Table S4. These molecules represent promising candidates for the development of pan-amyloid therapeutic agents and warrant further investigation through in vitro and in vivo validation studies.
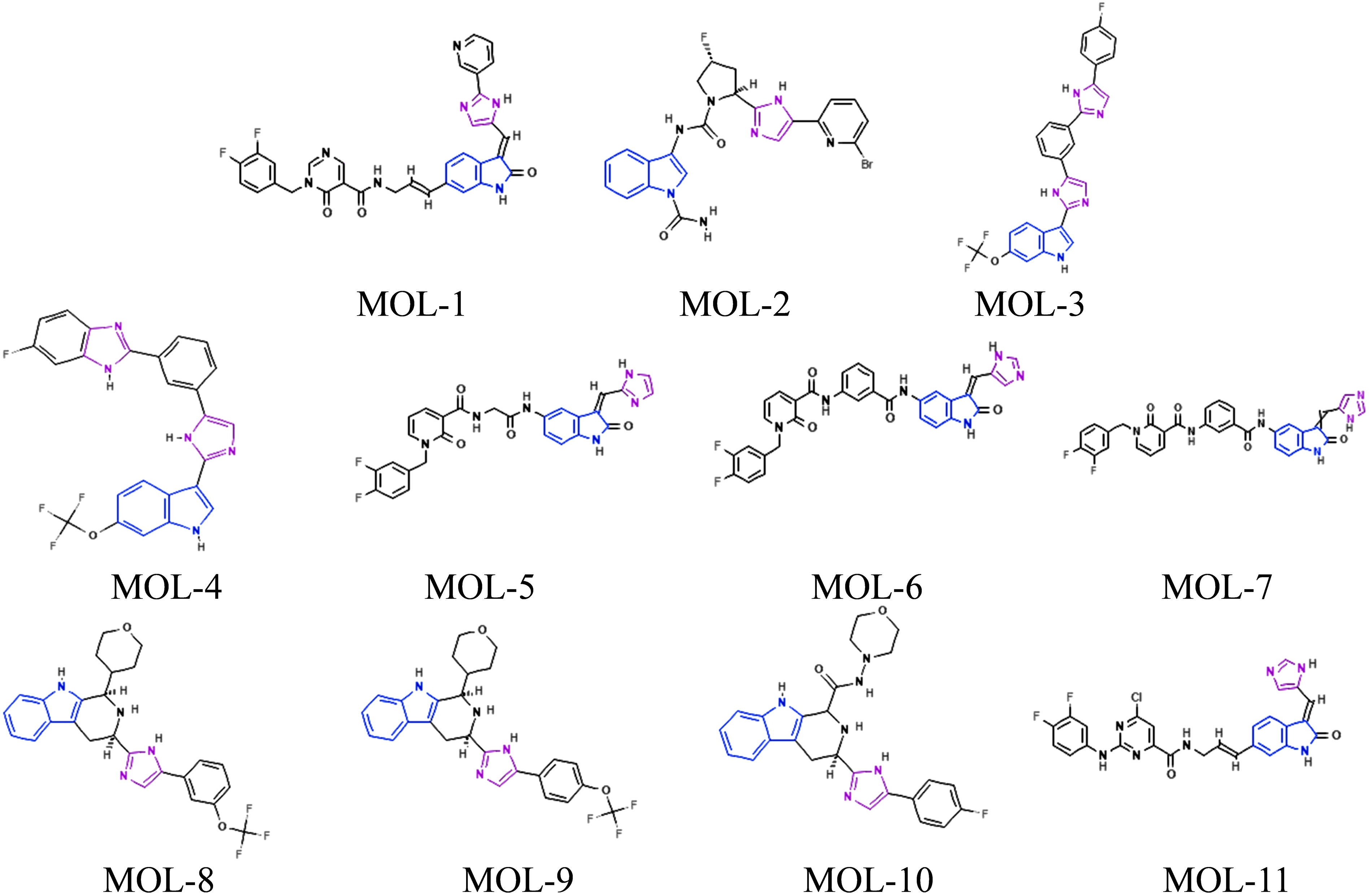
The searched 11 small molecules containing indole and imidazole moieties in the identified potential molecules.
Comparison of Binding Energy
We collected several known amyloidogenic inhibitors, including Tafamidis, 36 EGCG, 37 Curcumin, 38 Resveratrol, 39 and Doxycycline. 40 Using the same docking parameters, we docked these five molecules into four amyloidogenic proteins and compared their docking energies with those of the identified potential molecules. For convenience, the average docking energy of these 11 molecules was used for comparison with the five known inhibitors. The comparison results are shown in Figure 8A. For the four amyloidogenic proteins considered, the five known inhibitors exhibited comparable binding energies. However, compared to 11 molecules, the average docking energy of these molecules was higher than that of all five inhibitors, indicating that these 11 molecules have a stronger binding affinity to the amyloidogenic proteins than the five known inhibitors.
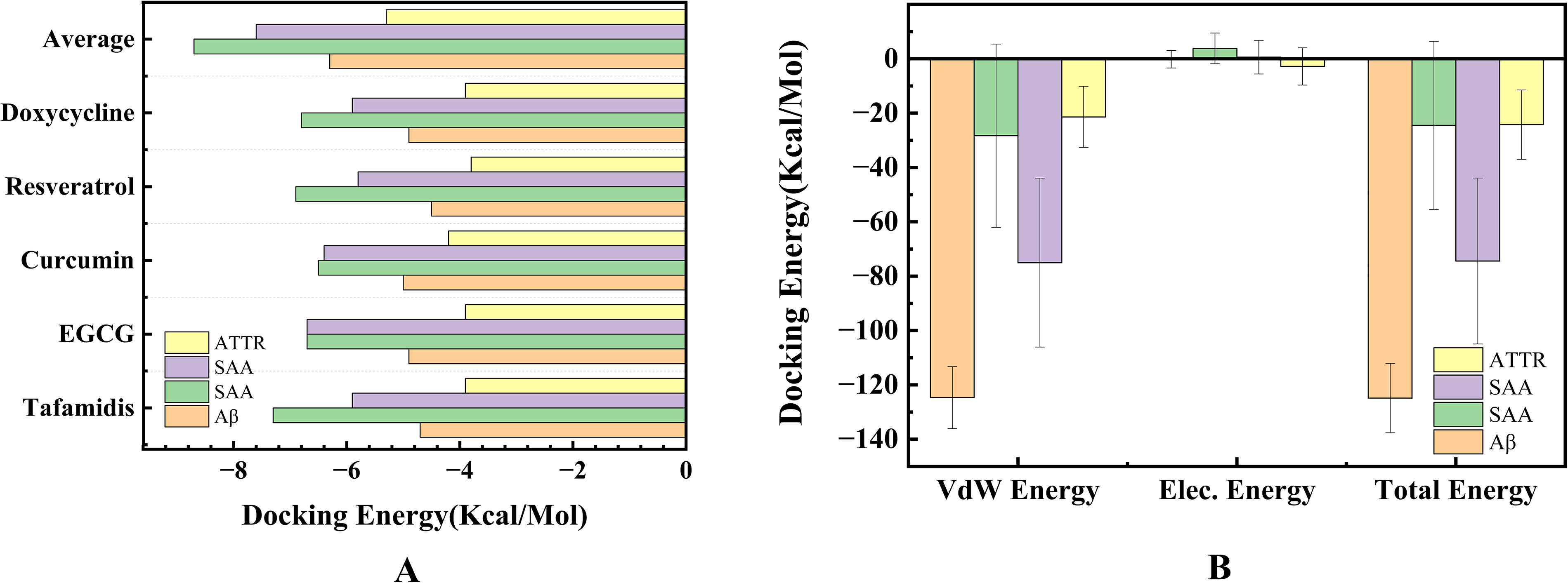
Comparison of docking energies between the average values of 11 molecules and those of five known inhibitors
Furthermore, we conducted MD simulations of Mol-1, which exhibited the highest binding energy among 11 molecules bound to four amyloidogenic proteins. Based on the MD trajectories, we calculated the binding energy of Mol-1 with the four amyloidogenic proteins using the MM-PBSA method. This approach allowed us to determine the total protein-ligand interaction energy, as well as the contributions of van der Waals (vdW) and electrostatic interactions, from the extracted the conformations. Figure 8B shows that the interaction energy with the amyloidogenic proteins is higher, with the vdW interaction being the most significant contributing term. This finding is consistent with the binding of COM1 and COM2, from which the functional groups were identified. The results comparing the binding energies with known inhibitors and the calculated binding energies from MD simulations validate the identified potential molecules based on functional group-driven screening.
CONCLUSIONS
This study presents a functional group-based virtual screening strategy that systematically identifies pan-amyloid inhibitors by focusing on conserved interaction groups. Through a combination of high-throughput docking, hierarchical library refinement, and conformational analysis, we establish key functional groups, with indole and imidazole recognized as two central pharmacophores driving multitarget amyloid engagement. Compared to conventional ligand-based approaches, our framework demonstrated enhanced binding affinity, thermodynamic stability, and structural convergence across diverse amyloid proteins. These findings not only provide new chemical leads but also a rational design paradigm for developing broad-spectrum therapeutics against amyloid protein diseases. Furthermore, this screening strategy can be expanded to other aggregation-prone protein systems.
AUTHORS’ CONTRIBUTIONS
J.H. wrote the article and performed the docking analysis. C.D. is responsible for data processing. S.Z. analyzed the docking results. X.Y. is responsible for designing the research scheme. M.Z. is responsible for docking analysis. T.Z. revised the article.
Footnotes
AUTHOR DISCLOSURE STATEMENT
No competing financial interests exist.
FUNDING INFORMATION
This research was funded by