
Abstract
Amelogenin, the primary protein of the enamel matrix, has long been implicated in regulating crystal nucleation, growth, and spatial organization during tooth development. This study investigates how the absence of amelogenin affects enamel structure and mineralization. Using amelogenin knockout mice, we examine its role in maintaining enamel integrity, modulating ameloblast vesicle dynamics, and facilitating calcium ion transport through specific channels to the enamel surface. The goal is to uncover the mechanistic contributions of amelogenin to enamel biomineralization and its broader implications for dental tissue engineering and pathology. Our study demonstrates that the absence of amelogenin leads to profound disruptions in enamel formation and mineral transport. In amelogenin-null mice, the typical enamel layer was absent and replaced by peg-like, tapered mineral structures. These pegs stained positively for calcium (via alizarin red) and inorganic phosphate (via von Kossa’s method), indicating aberrant mineral deposition. Electron diffraction revealed that the pegs contained bundles of thin, parallel-aligned crystals with patterns consistent with calcium hydroxyapatite, confirming their mineralized nature. At the cellular level, ameloblasts in wild-type mice displayed large, bilayered vesicles (∼200 nm in diameter) at their apical poles, containing inorganic phosphate as detected by modified submicroscopic von Kossa staining. In contrast, amelogenin-deficient ameloblasts lacked both the bilayer membrane structure and phosphate labeling within these vesicles, suggesting disrupted vesicular transport and ion packaging. Further, in vivo calcium labeling with Fluo-4 showed successful apical transport of calcium to the enamel surface in wild-type mice. However, in the absence of amelogenin, calcium was aberrantly retained at the basal ameloblast pole and in the stratum intermedium. This mislocalization correlated with altered expression and distribution of intracellular calcium channel proteins, as shown by immunoreactivity. Together, these findings expand the functional role of amelogenin beyond structural organization during early enamel crystal formation. They reveal a previously underappreciated role in mediating vesicle architecture, phosphate loading, and directional calcium ion transport essential for proper enamel mineralization.
Keywords
Introduction
Tooth enamel is the hardest and most mineralized tissue in the human body, forming a protective outer layer that is critical for lifelong oral function and health. Its formation during amelogenesis requires the seamless coordination of mineral ion transport, matrix protein secretion, and crystal growth, all orchestrated by a specialized layer of epithelial cells called ameloblasts. The demands placed upon the cellular environment during enamel formation are extraordinary: vast quantities of calcium and phosphate must traverse the ameloblasts without disrupting cellular homeostasis or triggering cytotoxic responses. Understanding the mechanisms that enable this biological feat is not only essential for unraveling normal tooth development, but also for identifying the causes of enamel pathologies and informing regenerative therapies. 1
Mammalian teeth are covered by an exceptionally thick layer of tooth enamel, often as thick as the underlying dentin. The formation of this exquisite hard tissue layer requires the transport of a substantial amount of mineral ions from the bloodstream across the ameloblasts layer and to the enamel surface. The transport of these ions and proteins throughout ameloblasts poses numerous logistic challenges as the high levels of calcium and phosphate required for enamel development would interfere with the intracellular ameloblast ion balance. Recent efforts to explain ameloblast ion transport have focused on anion exchangers and regulators such as cystic fibrosis transmembrane conductance regulator (CFTR) and anion exchanger 2 (AE2) 2 as well as calcium channels such as STIM1, ORAi1, and SLC24A4.3,4 Two ion channel-associated proteins are known to cause enamel defects in humans, stromal interaction molecule (STIM1) and calcium release-activated calcium modulator 1 (ORAi1). In addition, the sodium/potassium/calcium exchanger SLC24A4 encodes a sodium/potassium/calcium exchanger protein called NCKX4, which has shown to play an essential role in human enamel formation. 3
The ion channel-associated proteins STIM1 and ORAi1 function through store-operated calcium entry (SOCE) in response to a depletion of endoplasmic reticulum-Ca2+ stores. 5 STIM proteins work as ER Ca2+ sensors and Orai proteins act as store-operated channels. 6 SOCE channels operate through calcium-permeable channels, especially transient receptor potential canonical 1 (TRPC1), which is recruited in response to ORAi1-mediated Ca2+ signals. Activation of TRPC1/STIM channels requires functional ORAi1. 7 SOCE functions as a response to prolonged store depletion and resulting Ca2+ entry that is sustained for minutes to hours, driving a wide range of biological processes from minutes to hours, including motility, gene transcription, signaling, and secretion. 6
Calcium channels and ion exchangers are not the only molecules required for successful amelogenesis. Loss of the principal enamel protein amelogenin leads to severe enamel defects,8–10 and mutations of individual enamel protein amino acids cause enamel defects of various degrees of severity.11,12 On a microscopic level, secretory ameloblast vesicles at the apical ameloblast pole are filled with a matrix that greatly resembles the protein matrix surrounding the initial enamel crystals.13,14 While there is an impressive body of work supporting the role of calcium channels in enamel-directed calcium ion transport, there is little known about the mechanism by which other essential enamel ions such as phosphates are transported, and it is not clear how the enamel proteins essential for the establishment of the initial enamel protein matrix are transported toward the developing enamel surface.
In previous studies, we have resolved the complex amelogenin protein structure, including an N-terminal alpha helix, followed by a histidine-rich stretch of 310 helices, the polyproline-rich tripeptide repeat region and a C-terminal hydrophilic telopeptide, 15 pointing to the complex structure and function of the assembled molecule. The well-defined yet mobile and flexible structure of the polyproline stretch has led to the hypothesis that such proteins may function as mineral-binding domains, protein–protein docking domains, or internal molecular spacers to facilitate enamel crystallite growth and organization.11,15,16 We have conducted ultrastructural studies suggesting that amelogenins undergo a gradual transition in their path from secretory vesicle matrix to initial enamel matrix to act as apatite crystal nucleators and packaging components involved in crystal growth and maturation.14,17
To investigate the role of amelogenin in enamel ion transport, we conducted a comprehensive study to determine how its absence alters the movement and fate of mineral ions and matrix proteins within the enamel organ. Specifically, we analyzed the composition and structure of mineral precipitates formed in amelogenin-deficient conditions and tracked the intracellular and extracellular pathways of calcium and phosphate as they traverse the enamel-forming epithelium. These experiments uncovered unexpected functions for amelogenin beyond its structural contributions to enamel crystallization, revealing its involvement in stabilizing transport vesicles, facilitating ion delivery to the enamel surface, and maintaining ion channel distribution within ameloblasts. Collectively, our findings offer new insights into the molecular mechanisms that support enamel biomineralization and highlight amelogenin as a critical regulator of ion trafficking during amelogenesis.
Methods
Animal experiments
For the preparation of 3 days postnatal mouse molars, amelogenin null and wild-type mice were sacrificed according to University of Illinois at Chicago (UIC) animal care regulation. To ensure adherence to ethical standards and minimize animal suffering, all procedures involving mice were conducted in accordance with institutional and national guidelines for the care and use of laboratory animals. Mice were closely monitored throughout the study for any signs of distress or pain. No signs of pain were observed during routine handling or experimental procedures. Prior to euthanasia, animals were deeply anesthetized using isoflurane to ensure complete loss of consciousness and prevent any perception of discomfort. Euthanasia was performed in compliance with American Veterinary Medical Association guidelines as approved by the UIC Institutional Animal Care and Use Committee (IACUC protocol #005-152). Tissues from three mutant mice and three wild-type control mice were used for comparison. For Fluo-4 labeling, pregnant mice close to delivery age were injected. Molars were dissected from mandibles and immersed into paraformalde (for scanning electron microscopy [SEM]), Karnovsky’s fixative (for transmission electron microscopy [TEM]) as previously described 18 or immersed into 70% ethanol for whole mount staining. Paraffin sections were cut at 5 μm thickness as previously described. 19
Alizarin red whole mount stain
For this procedure, first molars from amelogenin mutant and wild-type mice were fixed in 70% ethanol and then processed as previously described. 20 Briefly, tooth organs were fixed in acetone overnight and stained in Alcian blue for 4 h, followed by Alizarin red stain for another 4 h. The incubation in the dye solutions was followed by 1% KOH overnight for clearing. Molar tissues were then incubated in 50% glycerol/1% KOH until the tissues were cleared. Specimens were stored in 100% glycerol.
Von Kossa staining
Molar teeth of amelogenin mutant and wild-type mice were sagittally cut and then embedded in Kulzer 7200 plastic, sawn into thin slices and parallelly ground. Thin sections were stained with 5% silver nitrate for 15 min and thereafter rinsed with distilled water.
Immunohistochemical staining
Paraffin sectioned slides were used for immunohistochemistry to detect STIM1, ORAi1, SLC24A4, SLC4A7, and CFTR proteins. Briefly, sections were incubated with rabbit or mouse polyclonal or monoclonal antibodies, followed by a biotinylated anti-rabbit or anti-mouse IgG secondary antibody (ImmPress Excel Amplified Polymer Kit, Peroxidase; Vector Laboratories, MP-7601 and MP-7602), according to the manufacturer’s instructions. Nuclei were counterstained with hematoxylin for 1 minute. At least three samples were analyzed. Control sections were incubated with nonimmune rabbit or mouse IgG of the same concentration.
Antibody details:
STIM1 Monoclonal Antibody, Invitrogen (Product # MA1-19451), RRID AB_2197884, 1:200 SLC24A4 Monoclonal Antibody, Proteintech (Product # 18992-1-AP), 1:200 SLC4A7 Polyclonal Antibody, Invitrogen (Product # PA5-104154), RRID AB_2853482, 1:200 CFTR Monoclonal Antibody, Invitrogen (Product # MA1-935), RRID AB_2081230, 1:200 ORAI1 Polyclonal Antibody, Invitrogen (Product # PA5-109270), RRID AB_2854681, 1:200
Scanning electron microscopy
For scanning electron microscopy, whole mouse molar teeth from wild-type and control animals were fixed in 4% paraformaldehyde for 4 h and etched using ethylenediaminetetraacetic acid (Warshawsky and Moore 1967). 21 Etched enamel surfaces were then analyzed using a JOEL JSM-6320F scanning electron microscope.
Transmission electron microscopy
Three days postnatal mouse molar tooth organs were fixed in Karnovsky’s fixative as previously described, 20 dehydrated and embedded in Eponate 12 (Ted Pella, Redding, CA). Sections were cut on a Leica Ultracut UCT 7 ultramicrotome at 80 nm thickness. After drying, sections were contrasted in 1% uranyl acetate followed by Reynold’s lead citrate for 15 min each. Observations were made on a Japan Electron Optics Laboratory Company 1220EX transmission electron microscope. Electron diffraction was performed on 120 nm thick sections positioned on uncoated copper grids. Spectra were obtained in the diffraction mode at 80 kV and a camera length of 80 cm. Measurements were made at a 90-degree incident to the samples. Patterns were measured for spot or ring diameter directly from the micrograph and d spacings were compared with those characteristic for hydroxyapatite.
Modified von Kossa stain for electron microscopy
Ultrathin sections of 80 nm thickness were incubated in phosphate-buffered saline overnight and then etched in 10% ammonium phosphate for 2 h followed by incubation in 5% silver nitrate for 2 min in sunlight or under a UV bulb. Thereafter, samples were transferred to a darkroom. In the darkroom, sections were developed in Kodak HC110 developer Dilution B for 6 min, followed by rinsing for 1 min in 10% acetic acid and another incubation in 10% sodium thiosulfate for 5 min. Thereafter, sections were briefly rinsed in distilled water and air-dried. At this point, samples were returned to a daylight room.
Fluo-4 injections and tissue preparation
Fluo-4 was dissolved in sterile 0.9% sodium chloride and DMSO. Pregnant mice carrying embryos of delivery age received i.p. injections using the dosages of 5 mg/kg body weight. Samples were harvested after 24 h Fluo-4 incubation and fixed with ice-cold 4% PFA overnight, embedded in frozen tissue embedding media (Fisher), then cryosectioned at 10 μm, and loaded on glass slides. The sections were rinsed with phosphate buffered saline, counterstained with 4′,6-diamidino-2-phenylindole, mounted with anti-fade mounting medium (Invitrogen) and examined using conventional fluorescence microscopy and laser-scanning confocal microscopy.
Results
Loss of amelogenin enamel protein was associated with the presence of calcium–phosphate-rich pegs replacing the enamel layer
Wildtype mouse first molars are adorned with seven cusps, organized into buccal, lingual/palatal and mesial/distal locations. Molar cusps were covered with a thick enamel layer, predominantly on distal slopes. Alizarin red and von Kossa staining reactions attest to the high calcium (alizarin) and phosphate (von Kossa) content of these tissues (Fig. 1). Wild-type mouse molars were compared to mutant counterparts in which the major tooth enamel protein, amelogenin, was absent due to genetic manipulation. In these mice, the enamel layer was replaced by irregular assemblies of pegs (pg) at the site of the enamel covering. These pg stained positively for alizarin red (calcium) and von Kossa (phosphate) as demonstrated by thin ground sections (Fig. 1).
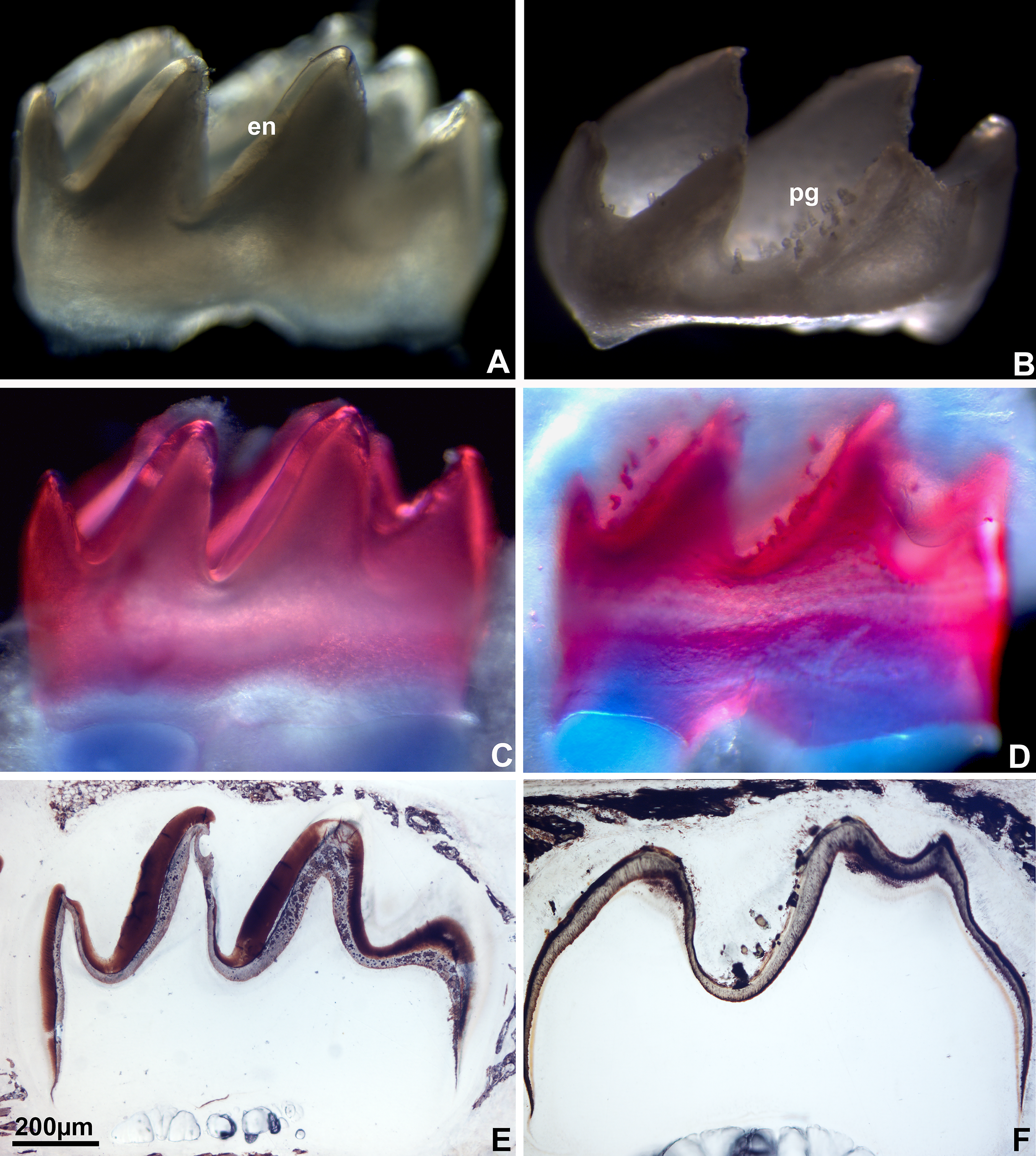
Enamel coverings in wild-type mice and enamel pegs (pg) of amelogenin null mice as visualized using light microscopy.
Mineral pg located on amelogenin null mouse molars were characterized by cylindrical tapered shapes
Following 2 weeks of incubation in Hank’s buffer, enamel organ tissues were gently removed using dissection tweezers (Fig. 2). Environmental SEM (ESEM) faithfully reproduced the original peg shape in nondried specimens (Fig. 2). Following desiccation, vacuum-based scanning electron microcopy imaged the pg and revealed the underlying crystal structure (Fig. 2).
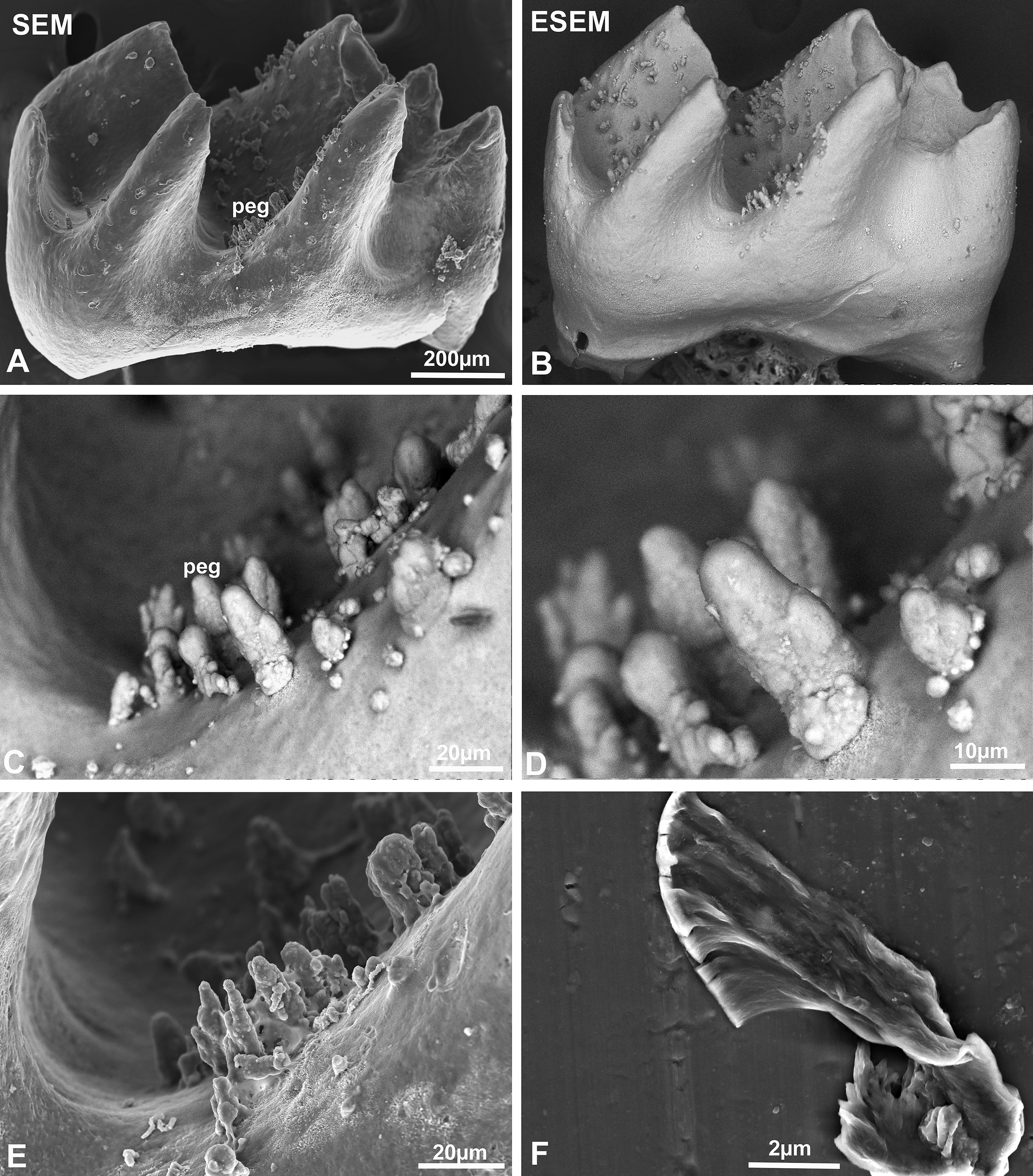
High magnification visualization of the mineral pegs on the surface of amelogenin null mouse molars using scanning electron microscopy.
Separated enamel organ pg were crystalline and contained calcium and phosphate
Upon separation from the enamel surface, mineral pg were retained in the enamel organ (Fig. 3). Pg were imaged using phase contrast and darkfield microscopy. Enamel organs of amelogenin null mice were separately embedded and probed with von Kossa’s stain for inorganic calcium, staining positive (Fig. 3). Transmission electron microscopic analysis of the ultrastructure of the mineralized pg revealed enamel-like crystal bundles or rows of mineralized platelets, indicative of initial crystal assemblies via Ostwald ripening (Fig. 3). Electron diffraction analysis confirmed the crystalline calcium content of these deposits via electron diffraction (Fig. 3).
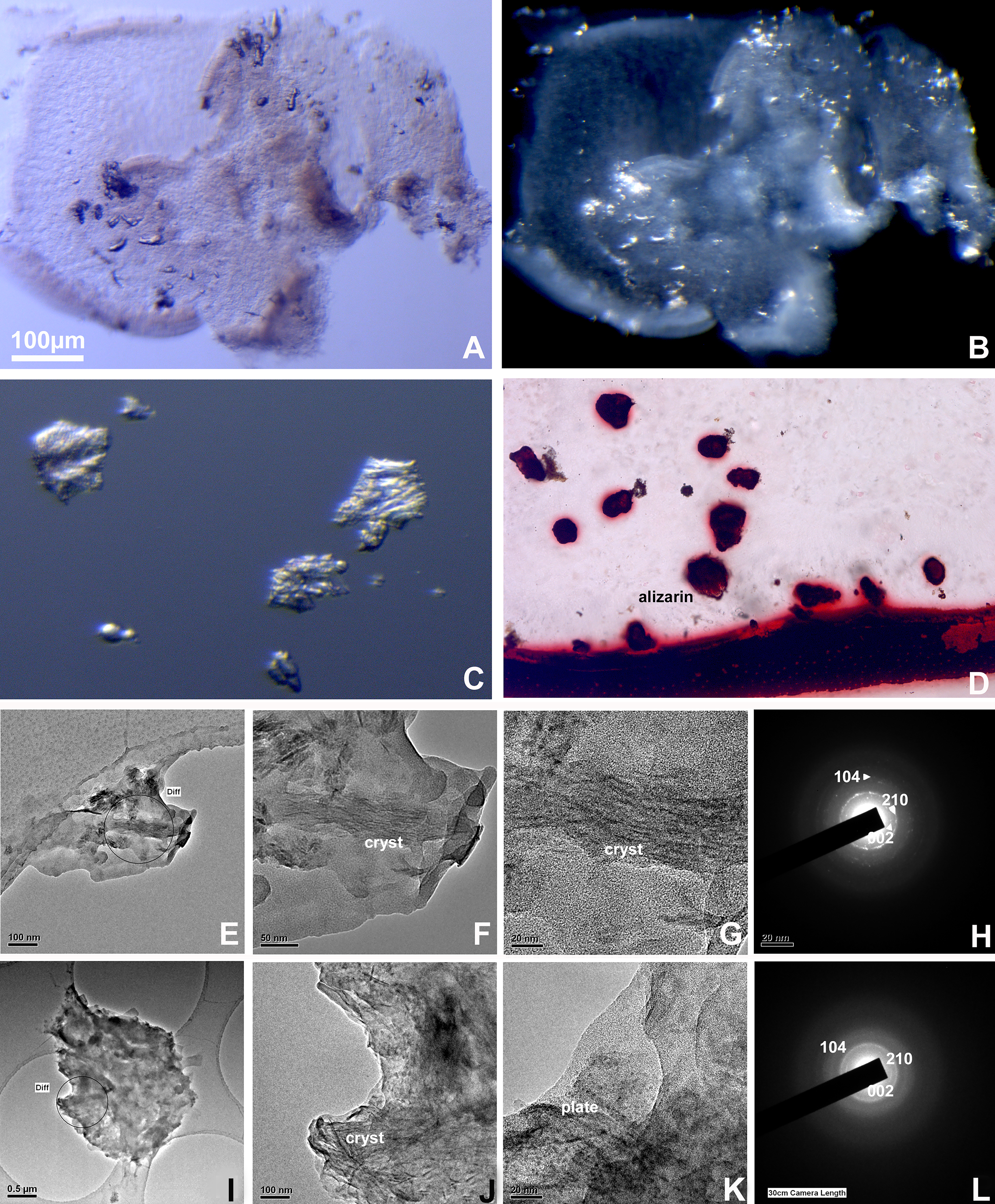
Analytical investigation of enamel organ-based pegs.
Large, amelogenin protein-containing vesicles were depleted of their inorganic phosphate content upon loss of amelogenin
Secretory ameloblasts from 3 days postnatal tooth organs prominently featured 100–200 nm diameter vesicles containing electron dense material and surrounded by a bilayer membrane (Fig. 4). In contrast, matching vesicles from amelogenin null mice were relatively electron lucent and lacked the bilayer membrane (Fig. 4). We developed a latent von Kossa stain for electron microscopy useful to identify mineralized phosphate. Densely packed and thick particles were revealed in the vesicles of wild-type mice while prominent phosphate-labeling precipitates were nearly absent from ameloblast vesicles in amelogenin null mice (Fig. 4). Immunoreactions for amelogenin highlighted amelogenin as a protein component of the secretory ameloblast vesicle walls (Fig. 4).
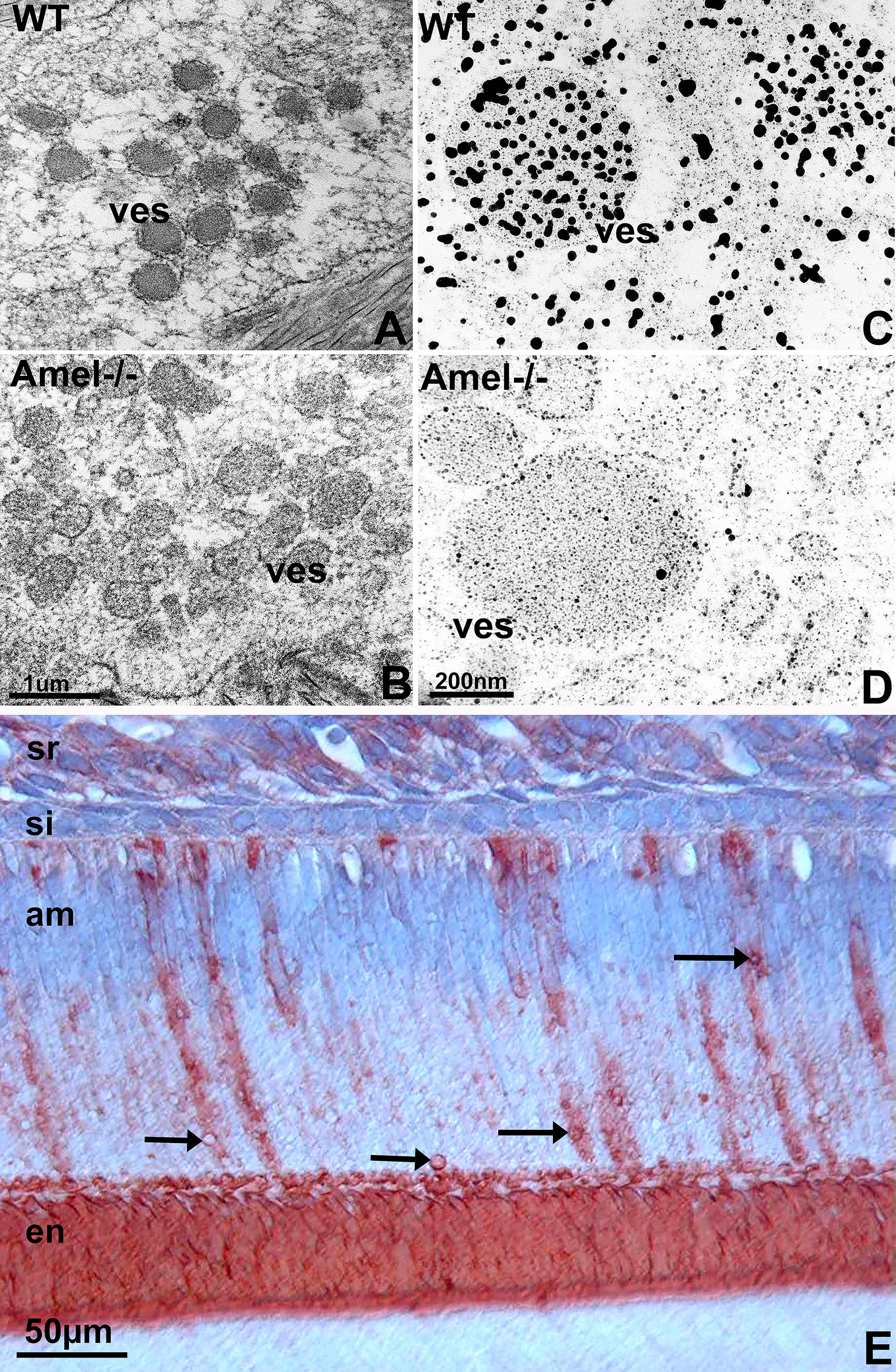
Loss of amelogenin impairs ameloblast cargo transport as visualized by high resolution modified von Kossa reaction.
Fluo-4 calcium labeling reached the enamel layer in wild-type mice following intraperitoneal dye injection, while fluo-4 was retained at the basal ameloblast pole of amelogenin null mice
For this study, the intracellular calcium ion indicator Fluo-4 was intraperitoneally injected and its distribution in the 3 days postnatal mouse molar tooth organ was examined using fluorescent microscopy following frozen section preparation. In wild-type tooth organs, the green Fluo-4 label was detected in the stratum intermedium, in the ameloblast, on the surface of the enamel layer, and in the odontoblasts (Fig. 5). In amelogenin mutant mice, the green Fluo-4 label was retained in the stratum intermedium and at the basal ameloblast pole, while no label was present at the secretory pole of the ameloblasts and at the enamel surface. The odontoblasts were stained both in wild-type and amelogenin-null tooth organs (Fig. 5).
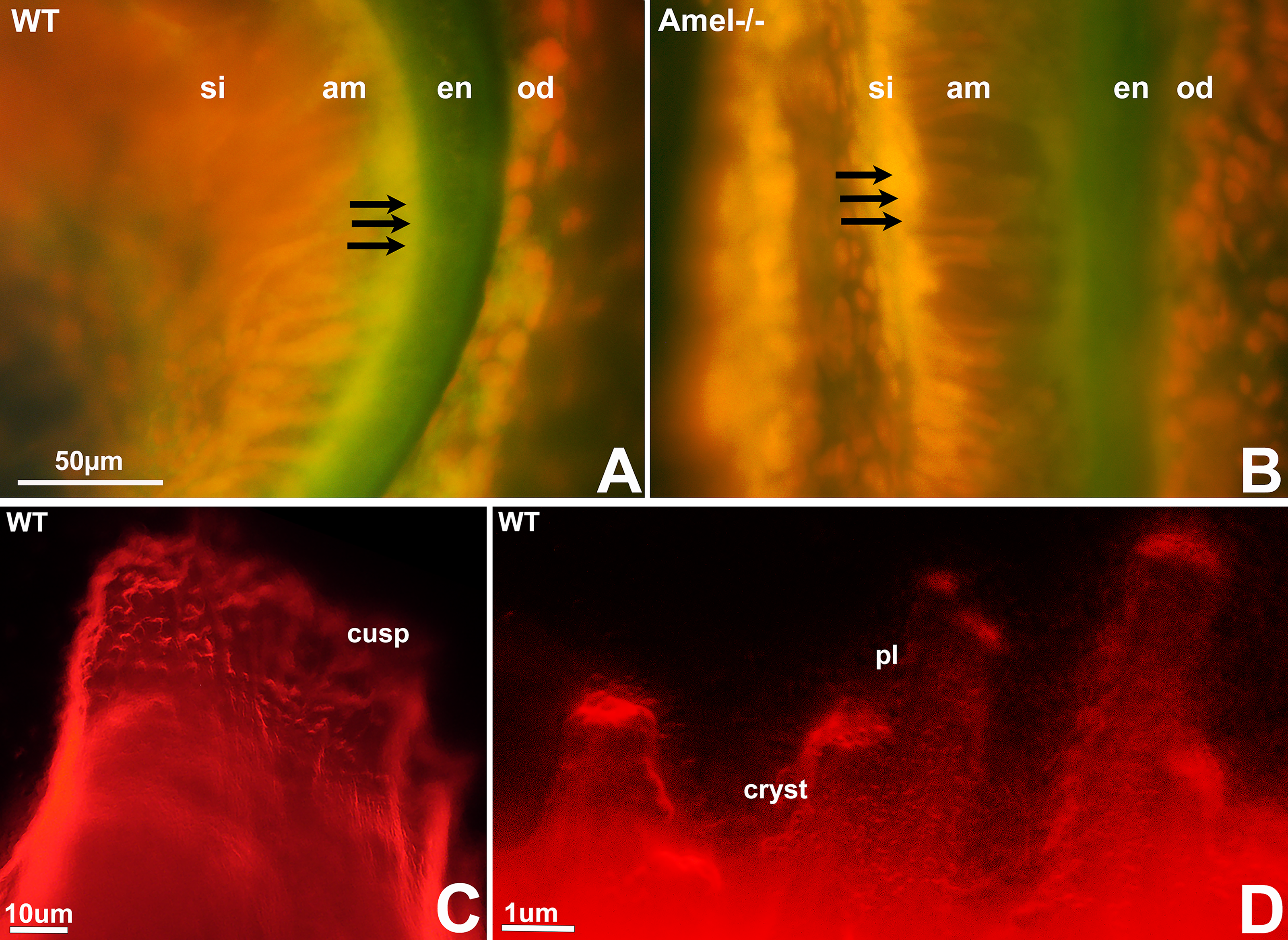
Failure of trans-ameloblast ion transport in amelogenin mutant mice.
Lack of amelogenin altered the unique subcellular distribution of ion channel proteins in wild-type mice
In the present study, we have compared the distribution of calcium ion channel proteins such as CFTR, ORAi1, SLC4A7, SLC24A4, and STIM1 in the ameloblast layer of wild-type and amelogenin-null mice. While all ion channel proteins, with the exception of CFTR, were widely distributed throughout the ameloblast layer
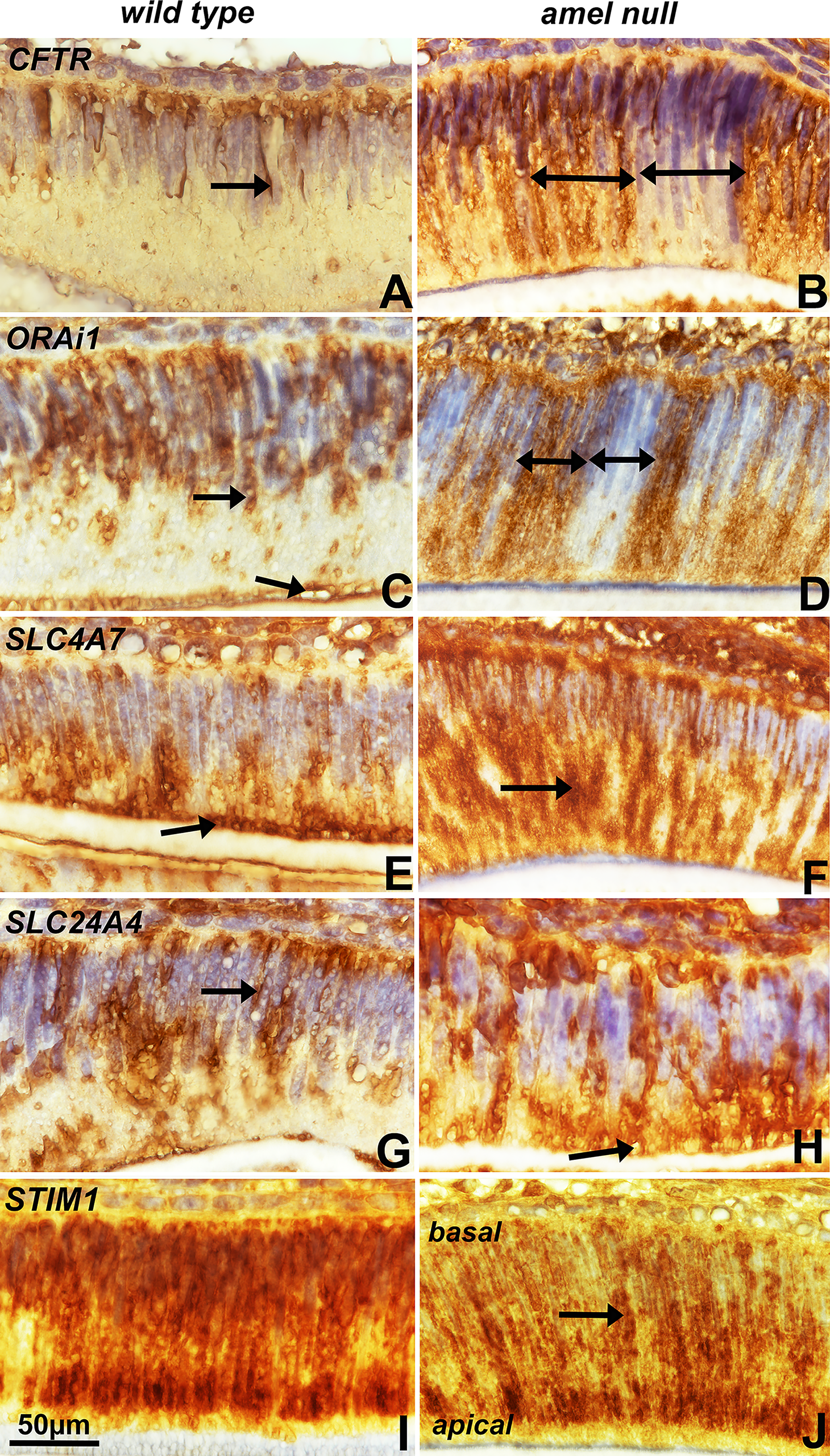
Immunolocalization of ion channel proteins in 3 days postnatal secretory ameloblasts. Prominent sites of immunostaining have been demarked by black arrows.
Discussion
This study presents the first compelling evidence that amelogenin, beyond its well-established role in organizing the developing enamel matrix, is also a critical regulator of ion transport during amelogenesis. Our findings reveal a previously underrecognized functional dimension of amelogenin, positioning it as a key mediator of vesicle structure, phosphate compartmentalization, and directional calcium transport across the ameloblast layer. These insights advance the current understanding of enamel biomineralization by highlighting the active, rather than solely structural, participation of amelogenin in the tightly regulated process of ion delivery essential for proper enamel crystal growth and maturation. From a technical perspective, the study has been based on 3-day postnatal first mandibular mouse molars as our standard model for the electron microscopy of enamel development, representing the most advanced age for routine undecalcified sections through developing enamel using a diamond knife.10,19,23 In contrast to wildtype molars, amelogenin null tooth organs featured prominent mineral pg throughout the stellate reticulum layer, at the site of the ameloblast layer and in the stratum intermedium. 24 Von Kossa staining and electron diffraction analysis revealed that pg were condensed protein-less mineral accumulations. To trace mineral ions on their path to the enamel layer, Fluo-4 labeling and a latent von Kossa procedure were applied. To examine the involvement of enamel organ ion channels, STIM and ORAiI were compared by immunostaining.
Exploring the fate of these extraneous ions in the enamel organ, we noted calcium and phosphate-rich pg in the enamel organ of amelogenin null mice. Others have reported ectopic mineral pg in the enamel organ of ameloblastin or enamelin mutant mice, suggesting that ameloblastin and enamelin also play a role in the ion transport throughout the ameloblast layer. Together, our findings of disrupted calcium transport and enamel organ-bound mineral pg demonstrate that amelogenin was necessary to facilitate the transport of mineral across the ameloblast cell layer. However, further studies may be needed to directly associate the amelogenin proteins with calcium phosphate ion and mineral transport.
Von Kossa’s reaction is a light microscopic staining procedure to label inorganic calcium on histologic sections. Here we have modified von Kossa’s reaction as a staining procedure for electron microscopy. To augment the silver phosphate grains of the original procedure, precipitates were developed in a photographic developer such as Kodak HC-110, washed in vinegar and fixed in sodium thiosulfate. Fixed and rinsed samples were dried and prepared for TEM. Wild-type controls revealed large-grain precipitates (20 nm versus 2 nm) demarking ameloblast secretory vesicles, while amelogenin null secretory vesicles did not yield large-grain precipitates and also lacked a bilayer membrane surrounding secretory vesicles and characteristic of their wild-type counterparts. Together, these data demonstrate that amelogenins are involved in phosphate transport across ameloblasts.
Our marker studies using the cell-permeable calcium marker Fluo-4 demonstrated that in 3 days postnatal wild-type mice, Fluo-4 had reached the developing enamel layer in 24 h interval samples. In contrast, in amelogenin null mice, Fluo-4 was absent from the enamel layer and secretory ameloblasts, while there was an intense staining in the stratum intermedium and at the basal pole of the ameloblast layer. These data indicate that in amelogenin null mice, calcium ion transport directed toward the enamel layer was disrupted prior to entering the ameloblast/stratum intermedium boundary.
Our immunoreactions demonstrated that important ion channels were localized in the secretory ameloblast layer and that loss of amelogenin resulted in a changed distribution pattern for these ion channel proteins. Especially CFTR, ORAi1, SLC4A7 and SLC24A4 were increased in amelogenin null mice, while STIM1 was increased in wild type mice when compared to amelogenin null mice. This study demonstrates that amelogenin affected the distribution of ion channels in ameloblasts. There is ample support for the role of calcium channels across the ameloblast barrier and directly onto the forming enamel surface. This includes findings of severe enamel hypomaturation in patients with genetic mutations in the ORAi1 transmembrane protein and Ca2+ influx regulator, 25 of the STIM1 transmembrane protein, 4 and of the potassium-dependent sodium-calcium exchanger SLC24A4. 26 Together, defects in ORAi1 and STIM1 abolish CRAC channel function as they relate to SOCE calcium stores. 27 However, further studies have associated STIM1 and ORAi1 with maturation stage rather than secretory ameloblasts5,28 and it is not clear whether STIM1 and ORAi1 are specifically related to the secretion and transport of enamel-related minerals as they play important roles in many non-ameloblast cells. However, ions transporters such as SCL24 hold great potential as intermediaries between calcium ions channels and membrane regions on the path of ions toward the developing enamel layer.
So far, our studies have characterized amelogenin as a versatile molecule involved in multiple aspects of ion and protein transport during amelogenesis. On a protein level, secretory vesicles transport amelogenins as a finely stippled matrix component at the secretory (apical) ameloblast pole, followed by teleopeptide cleavage and expansion of the matrix subunit dimensions upon entry into the extracellular space. Our staining data indicate that phosphate ions travel together with the secretory vesicles to the enamel crystal nucleation site. Little is known about the fate and pathway of other enamel ions, especially Ca2+, the most prominent component of the inorganic enamel layer. Multiple studies, including the present one, indicate that calcium channels contribute to amelogenesis. However, it is not clear whether calcium and phosphate travel together through the ameloblast layer or whether they are separated from each other during transport by embarking upon different pathways (eg, vesicle versus channel) or whether they reside in different subunit compartments of the forming enamel matrix.
In bone, matrix vesicles carry calcium and phosphate ions embedded in collagen matrix proteins directly to the mineral nucleation site. However, enamel is not bone, and the enamel-specific matrix protein amelogenin is distinctly different from the ubiquitous bone matrix protein collagen. At this point, the science of ion transport to the enamel mineral layer remains exciting and wide open.
Footnotes
Acknowledgment
Fluo-4F labeling studies were performed by Yoshihiro Ito.
Author Disclosure Statement
The authors have no conflicts to declare.
Funding Information
Funding from NIDCR grant UG3/UH3-DE028869 for this project is gratefully acknowledged.