
Abstract
In pancreatic cancer, increased collagen I impairs the efficacy of gemcitabine; however, the role of gemcitabine itself in collagen I accumulation remains unclear. This study aims to explore the mechanism of gemcitabine-induced fibrosis and provide new insights to enhance its therapeutic efficacy. We analyzed COL1A1 expression in pancreatic cancer patient tumor tissues and found that gemcitabine treatment upregulated COL1A1 expression. Subsequently, cancer-associated fibroblasts (CAFs) were modeled by inducing human adipose-derived mesenchymal stem cells with tumor-derived exosomes. Using the autophagy inhibitor chloroquine (CQ) and the protein kinase B (AKT) activator SC79, we demonstrated that gemcitabine downregulated P62 expression and upregulated LC3BII, Beclin-1 expression, inducing autophagy in CAFs via decreasing AKT phosphorylation, which further led to collagen I accumulation. In addition, gemcitabine combined with CQ enhanced cell death in both CAFs and tumor cells, while inhibiting tumor cell proliferation and migration. In animal models, this combination therapy reduced gemcitabine-induced autophagy and collagen I deposition, contributing to delayed tumor growth. Collectively, gemcitabine upregulates collagen I by inducing CAF autophagy via reducing AKT phosphorylation. Targeting CAF autophagy can reduce collagen deposition, offering a promising strategy to improve the therapeutic efficacy of gemcitabine in pancreatic cancer.
Introduction
Pancreatic ductal adenocarcinoma (PDAC), one of the most aggressive malignancies, is routinely treated with gemcitabine as a first-line chemotherapeutic agent. 1 Emerging evidence implicates excessive collagen deposition as one of the key contributors to PDAC chemoresistance.2,3 Mechanistically, dense collagen networks not only promote tumor cell growth and migration 4 but also form a physical barrier affecting drug delivery, thereby compromising therapeutic efficacy. Collagen I is associated with tumor cell proliferation and epithelial–mesenchymal transition 5 and may affect M2 macrophage infiltration, 6 all of which indirectly impair the efficacy of gemcitabine. Therefore, strategies aimed to enhance chemotherapeutic efficacy by overcoming collagen-mediated drug resistance have become a key focus in current PDAC research.
Collagen I is the predominant form of collagen in the extracellular matrix, accounting for over 90% of total collagen content. 7 Cancer-associated fibroblasts (CAFs) are the most prevalent stromal cells in the tumor microenvironment 8 and play a pivotal role in promoting collagen deposition within the tumor extracellular matrix. 9 Furthermore, studies have identified pancreatic stellate cells and fibroblasts as the primary sources of collagen production in the pancreas. 10 Together, these findings indicate that CAFs are closely associated with increased collagen I deposition. Meanwhile, studies have confirmed that gemcitabine treatment increases collagen deposition in tumors.11,12 Gemcitabine-induced collagen I deposition may result from the combined effects of reactive fibrosis and a specific stress response in CAFs. However, the mechanism by which gemcitabine induces collagen I deposition and its relationship with CAFs remains unclear.
Proline is a vital amino acid component of collagen. Studies have demonstrated that autophagy can promote proline synthesis by regulating mitochondrial NAD kinase 2, and inhibiting autophagy in CAFs suppresses proline biosynthesis and reduces collagen production, 13 highlighting a critical link between CAF autophagy and collagen regulation. Similarly, Zhang et al. found that autophagic activity in CAFs was significantly correlated with collagen deposition in human PDAC tissues. 14 Other studies have reported that inhibiting autophagy reduces pancreatic fibrosis, decreases COL1A1 expression, and attenuates tissue collagen deposition. 15
Autophagy is a lysosome-mediated self-degradation process 16 that helps maintain cellular homeostasis. Interestingly, gemcitabine can induce autophagy in pancreatic cancer cells17,18; however, few studies have investigated the relationship between gemcitabine and autophagy in CAFs.
Given that CAFs regulate fibrogenesis through autophagy-dependent pathways, we hypothesized that gemcitabine triggers CAF autophagy to drive collagen accumulation. Therefore, this study aimed to explore the mechanism of gemcitabine-induced collagen I production in pancreatic CAFs and to evaluate whether targeting autophagy can abrogate this response, potentially providing a novel strategy to overcome chemoresistance in PDAC.
Materials and Methods
Patient samples
We collected needle biopsy and surgical specimens before and after neoadjuvant chemotherapy from pancreatic cancer patients who received gemcitabine-containing chemotherapy regimens at Peking Union Medical College Hospital (PUMCH). The clinical data of the eight patients are shown in Supplementary Table S1. All specimens were immediately formalin-fixed paraffin-embedded (FFPE) and stored properly. These FFPE specimens were used for immunohistochemistry (IHC). This study was approved by the Ethics Committee of PUMCH (I-23PJ491). Written informed consent was obtained from all participating patients prior to sample collection. The study was conducted in accordance with the Declaration of Helsinki. All patients were histologically diagnosed with pancreatic cancer by two independent pathologists from PUMCH, who also reviewed the slides to confirm the CAF-rich areas in the tumor tissues.
Cell culture
Pancreatic cancer cell lines (PANC-1, AsPC-1, PAN 02) were purchased from the Cell Resource Center at the Chinese Academy of Medical Sciences and cultured in high-glucose Dulbecco’s modified Eagle’s medium (Gibco, USA). All media were supplemented with 10% fetal bovine serum (Gibco, USA), penicillin (100 U/mL), and streptomycin (100 μg/mL), and all the cells were cultured at 37°C in a humidified incubator with 5% CO2. Discarded fat tissue was obtained from patients who underwent liposuction procedures. The human adipose-derived mesenchymal stem cells (AD-MSCs) were isolated and cultured as described in our previous study. 19 Mouse AD-MSCs were purchased from IMMOCELL (IM-M067). AD-MSCs used in this study were at passage 4 or lower.
Differentiation of human AD-MSCs into CAFs
After the AD-MSCs adhered to the 6-well plate and reached 70%–80% confluency, the medium was replaced with serum-free D/F12 medium (Gibco, USA). One hundred micrograms of tumor cell-derived exosomes were added to each well. The medium was replaced, and new exosomes were added every 48 h. Proteins were collected at days 0, 1, 3, 5, 7, and 9, or drugs were added for subsequent experiments.20,21 In this study, the term “CAFs” refers to CAFs differentiated from human AD-MSCs, rather than primary CAFs isolated from human pancreatic tumor tissues.
Western blot
Total protein was extracted using radio immunoprecipitation assay (RIPA) lysis buffer supplemented with 1% protease and phosphatase inhibitor cocktails (Solarbio, China). Protein concentrations were determined using a BCA protein assay kit (Beyotime, China). Approximately 15 µg of protein per sample was separated by 6%–15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride (PVDF) membranes. Membranes were blocked with 5% nonfat milk for 1 h at room temperature, followed by overnight incubation at 4°C with the following primary antibodies: glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (#5174, Cell Signaling Technology [CST], USA), CD63 (TS63; Invitrogen, USA), HSP70 (#4873, CST), HSP90 (#4877, CST), AKT (#4691, CST), and p-AKT (#4060, CST), P62 (#5114, CST), beclin-1 (BECN1; #3495, CST), LC3B (#43566, CST), Bcl-2 (#4223, CST), Bax (#5023, CST), and COL1A1 (#72026, CST). After washing with TBST, membranes were incubated with the second antibodies, including antirabbit IgG (#7074, CST) and antimouse IgG (#7076, CST) for 1 h. Protein bands were visualized using ECL reagent and quantified via densitometry using ImageJ software.
For all western blot experiments, band intensities were quantified using ImageJ software, normalized to the corresponding internal reference (GAPDH), and presented as relative protein expression. The full quantitative results for all western blot panels in the main figures are compiled in Supplementary Figure S1.
IHC and TUNEL assays
IHC staining was performed using a standard protocol as described previously. 22 TUNEL assay was used to detect apoptosis in tumor tissues according to the manufacturer’s instructions (Servicebio, China). The primary antibodies and the dilutions used in IHC staining were anti-COL1A1 (CST, #72026; 1:200), anti-P62 (Servicebio, GB11531; 1:500), and anti-BECN1 (Servicebio, GB115741; 1:500). The secondary antibodies used were HRP-conjugated goat antimouse IgG (Servicebio, GB23301; 1:200) and HRP-conjugated goat antirabbit IgG (Servicebio, GB23303; 1:200).
Animal model
C57BL/6N mice were subcutaneously injected with 5 × 106 pancreatic cancer PAN02 cells and 1 × 106 mouse AD-MSCs per mouse into the right forelimb axilla to establish subcutaneous tumor models. When tumors reached 100 mm3, mice were divided into three groups (8 mice/group): control, gemcitabine, and combination group. The control group received an intraperitoneal injection of equal-volume saline; the gemcitabine group received 50 mg/kg gemcitabine twice weekly; the combination group received 50 mg/kg gemcitabine twice weekly plus 80 mg/kg chloroquine (CQ) daily (all intraperitoneal). Tumor length/width was measured every 3–4 days during treatment. After 2 weeks of treatment, mice were observed for another 1 week (without treatment), then sacrificed to collect subcutaneous tumors and livers.
Statistical analysis
All data are presented as the mean ± standard deviation from three independent biological experiments. Statistical analyses were performed using GraphPad Prism (version 9.0). Student’s t-test was used to compare statistical significance between two groups. For comparisons among three or more groups, one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was applied. Spearman’s correlation analysis (with correlation coefficient r and P value reported) was used for gene–gene correlations. The Kaplan–Meier method and log-rank test were used to analyze survival time between different groups. A P value <0.05 was considered statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001).
Results
Gemcitabine treatment is associated with an increasing trend in COL1A1 expression in pancreatic cancer tissues
We collected eight pairs of specimens, each including a preneoadjuvant therapy biopsy specimen from pancreatic cancer patients and a postoperative specimen from the same patient, all of whom received gemcitabine-based neoadjuvant regimen. IHC reveals an increasing trend in COL1A1 expression in pancreatic cancer tissues after gemcitabine treatment (Fig. 1A). Further analysis using the KM-plotter online tool (https://kmplot.com/analysis/), which utilizes log2-transformed TPM gene expression data from GEO, EGA, and TCGA databases with standardized batch effect correction, shows that high COL1A1 expression is associated with poor overall survival (OS) in pancreatic cancer patients (Fig. 1B). Notably, Cancer Therapeutics Response Portal (CTRP) analysis shows a positive correlation between COL1A1 mRNA expression and gemcitabine resistance (Fig. 1C). Collectively, these findings suggest that gemcitabine-induced collagen deposition may be associated with reduced sensitivity to gemcitabine. Based on the structural characteristics of collagen I, we infer that collagen deposition functions predominantly as a mechanical barrier that limits drug penetration, thereby contributing to gemcitabine resistance.
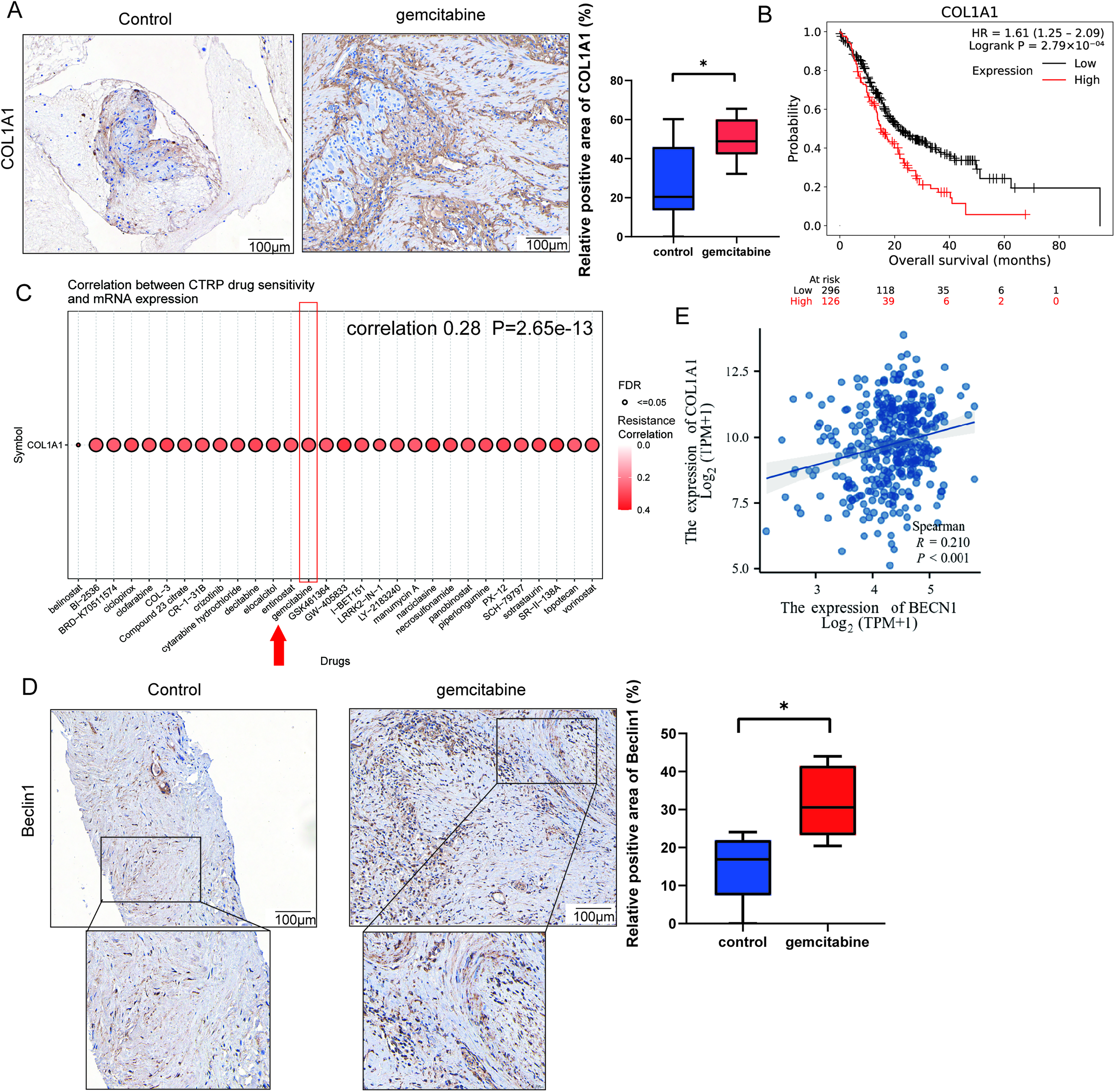
The expression of COL1A1 and Beclin1 after gemcitabine treatment in pancreatic cancer.
Gemcitabine-induced collagen I accumulation may correlate with CAF autophagy in PDAC specimens
Given that collagen in the tumor microenvironment is mainly secreted by CAFs,23,24 and emerging research suggests a correlation between increased CAF autophagy and collagen accumulation, 13 we further explored the role of autophagy in this process. Of the eight patients, three exhibited decreased autophagy after gemcitabine treatment. Among these three patients, one also showed decreased COL1A1 expression. To focus on samples with increased autophagy, we performed statistical analysis on the other five patients who showed an upward trend in BECN1 expression (Fig. 1D). In addition, analysis of the TCGA database indicates a significant positive correlation between the expression of COL1A1 and BECN1 (Fig. 1E). These findings suggest that gemcitabine-induced collagen I accumulation may be partially linked to autophagy. Owing to the small sample size and high heterogeneity in this clinical cohort, we subsequently performed in vitro and animal experiments to further verify the relationship between CAF autophagy and collagen production.
Pancreatic cancer cell-derived exosomes induce the differentiation of human AD-MSCs into CAFs
We extracted exosomes from pancreatic cancer cells and induced human AD-MSCs into CAFs for subsequent experiments.
Exosomes were isolated from pancreatic cancer cell lines PANC-1 and AsPC-1 using ultracentrifugation and subsequently characterized. TEM reveals the presence of exosomes with a bilayer membrane structure and a size range of 40–200 nm (Fig. 2A). The expression of exosome marker proteins, including HSP90, HSP70, and CD63, was detected. Besides, exosomes do not express calnexin in contrast to tumor cells (Fig. 2B). We isolated human AD-MSCs from adipose tissue and characterized them phenotypically via flow cytometry (Fig. 2C). Flow cytometric analysis confirms that the isolated AD-MSCs exhibit a characteristic surface marker profile, showing high expression (≥95%) of CD29, CD44, CD73, and CD90, while remaining negative (<2%) for hematopoietic and endothelial markers, including CD31, CD34, CD106, and HLA-DR, consistent with the defining surface marker profile of AD-MSCs.
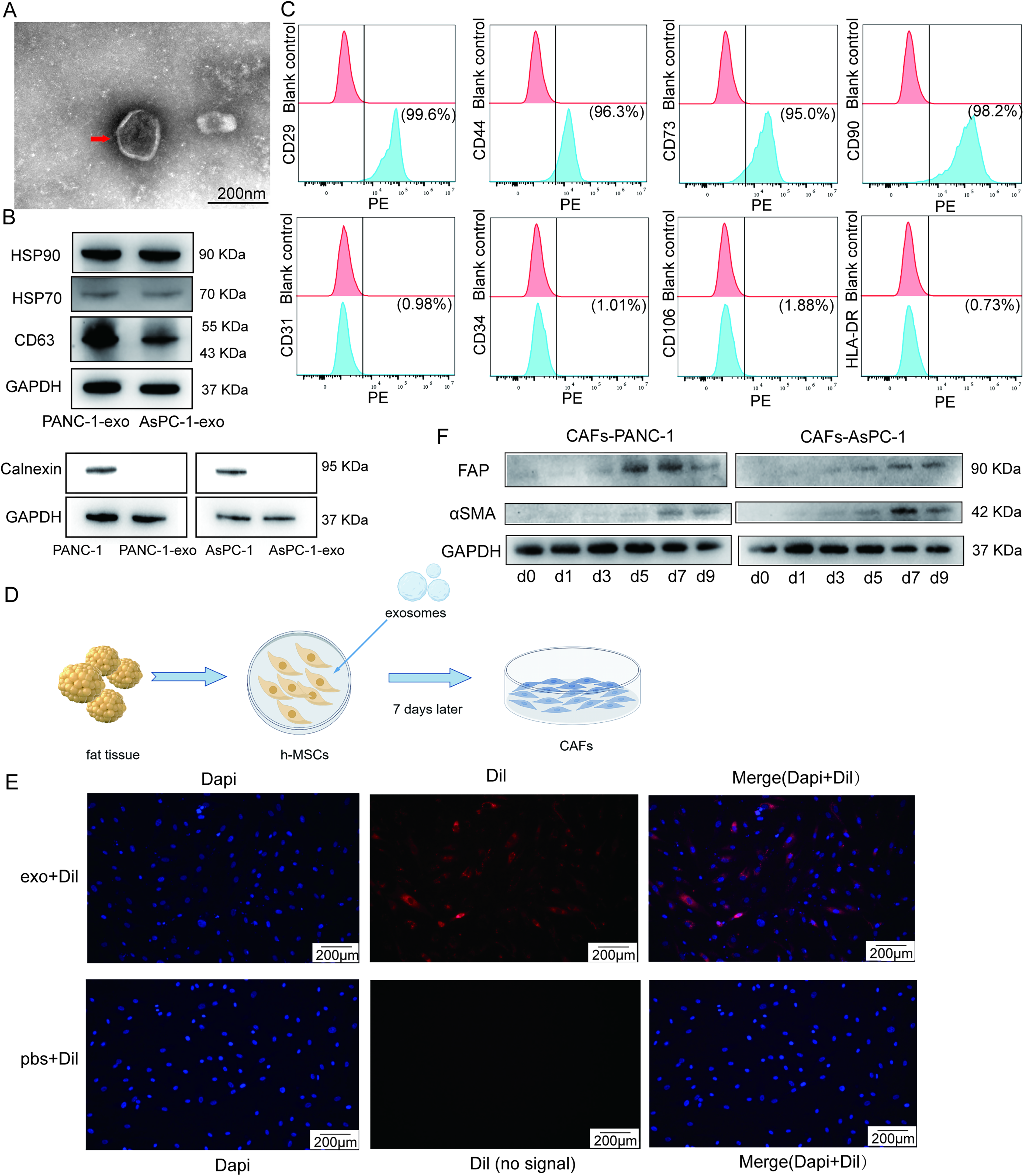
Pancreatic cancer cell-derived exosomes can induce the differentiation of human AD-MSCs into CAFs.
The process of inducing CAFs using exosomes derived from pancreatic cancer cells is illustrated in Figure 2D. To confirm the uptake of these exosomes by human AD-MSCs, AD-MSCs were incubated with Dil-labeled exosomes (red) and counterstained with 4',6-diamidino-2-phenylindole (DAPI) (blue) to visualize nuclei via fluorescence microscopy. Compared with the PBS control group, red dye is observed in the cytoplasm of human AD-MSCs in the exosome group, which shows AD-MSCs could uptake the exosomes of pancreatic cancer cells into the cytoplasm (Fig. 2E). Subsequently, we examined marker proteins of CAFs at days 0–9. The expression of CAF markers, including fibroblast activation protein (FAP) and alpha-smooth muscle actin (α-SMA), is highest on day 7 (Fig. 2F). Therefore, exosome-induced day 7 CAFs were used in all subsequent experiments.
Gemcitabine induces autophagy in pancreatic CAFs and increases collagen I
Current researches have reported that gemcitabine does not induce significant cell death in CAFs.25,26 Similarly, our study also found that the cytotoxic effect of gemcitabine at different concentrations on CAFs was not significant, which was consistent with current researches. Therefore, we selected the IC50 of the corresponding pancreatic cancer cells for subsequent experiments (Fig. 3A).
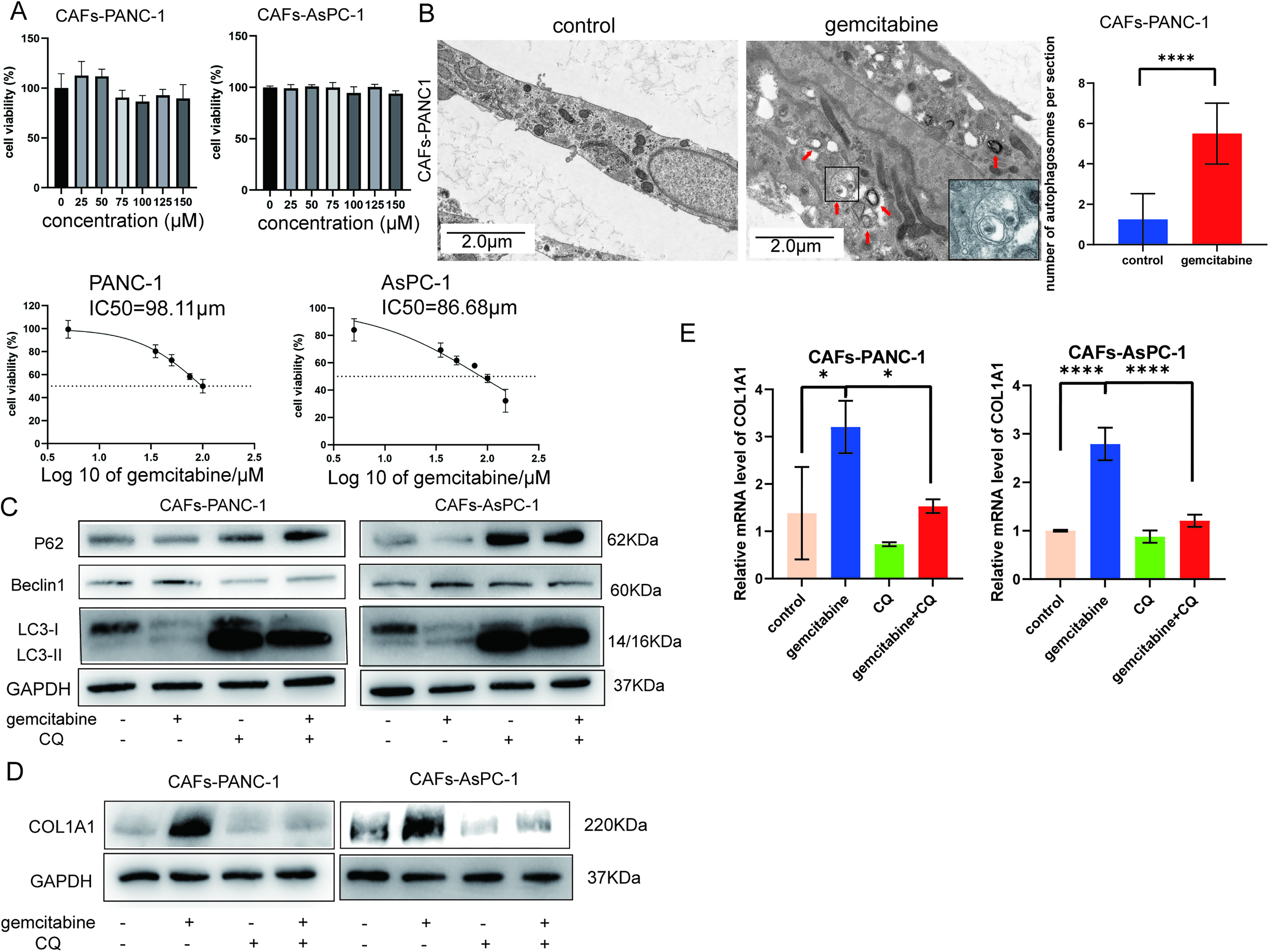
Gemcitabine induces autophagy and increases collagen I in CAFs.
Electron microscopy reveals double-membrane autophagosomal structures after gemcitabine treatment (Fig. 3B). Moreover, compared with the control group, the gemcitabine group shows increased beclin1 and LC3B, along with a decrease in P62, indicating that gemcitabine induces autophagy in CAFs (Fig. 3C). Notably, gemcitabine treatment increases COL1A1 expression in CAFs, whereas autophagy blockade with CQ reduces COL1A1 levels (Fig. 3D). The same results are observed at the mRNA level of COL1A1 (Fig. 3E).
Thus, we hypothesized that gemcitabine increases collagen I at least partially by inducing autophagy in CAFs.
Gemcitabine reduces the phosphorylation level of AKT in CAFs, inducing autophagy and collagen I accumulation
Given that studies have demonstrated an association between inhibition of AKT-related pathways and autophagy, 27 we performed the protein–protein interactions between AKT, autophagy and COL1A1 (Fig. 4A), and detected the correlation between COL1A1 and AKT pathway inhibition in the TCGA-PAAD database (Fig. 4B). To investigate whether the AKT pathway was involved in gemcitabine-induced autophagy in CAFs, we examined AKT phosphorylation (p-AKT) in CAFs and found that AKT phosphorylation was decreased after gemcitabine treatment (Fig. 4C).
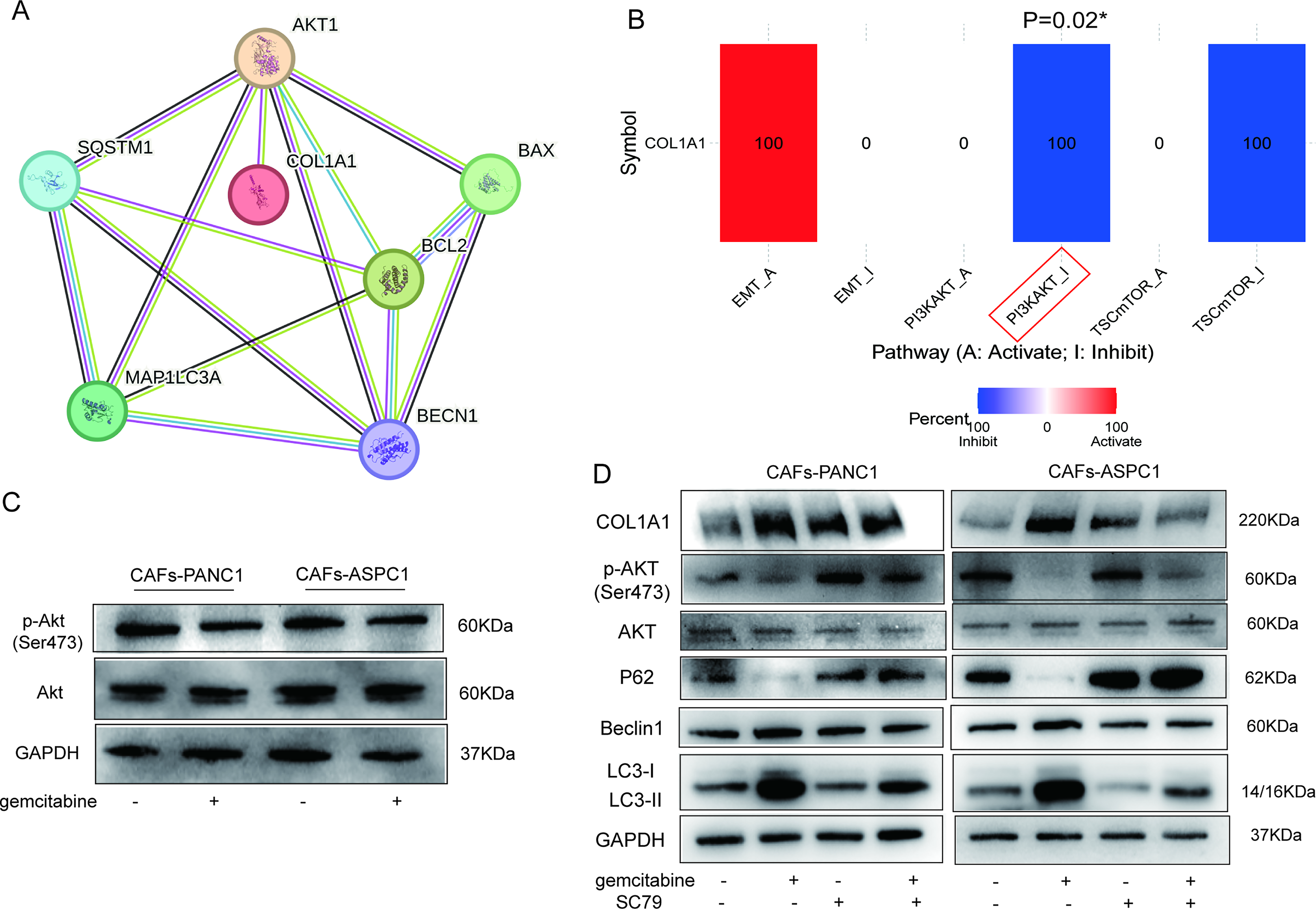
Gemcitabine induces autophagy in CAFs by downregulating AKT phosphorylation, leading to collagen I accumulation.
To further verify our hypothesis, we performed a rescue experiment using the AKT activator SC79 (10 μM) in combination with gemcitabine (Fig. 4D). Western blotting shows that cotreatment with SC79 reverses gemcitabine-induced autophagy, as evidenced by downregulated beclin1 and LC3BII expression and upregulated P62 expression in the combination group compared with the gemcitabine-treated group. Meanwhile, SC79 cotreatment partially reverses gemcitabine-induced upregulation of COL1A1. Notably, SC79 treatment alone does not significantly reduce baseline COL1A1 levels. These findings collectively demonstrate that gemcitabine decreases AKT phosphorylation in CAFs, thereby inducing autophagy and leading to collagen I accumulation.
Combination with CQ enhances the cytotoxicity of gemcitabine against CAFs and pancreatic cancer cells
Next, we targeted autophagy in CAFs to determine whether inhibiting CAF autophagy and reducing collagen I could enhance the sensitivity of pancreatic cancer cells to gemcitabine. First, a Transwell coculture system with 0.4 μm pores was established to model the interaction between pancreatic cancer cells and pancreatic CAFs. CAFs were seeded in the upper chamber, and pancreatic cancer cells were plated in the lower chamber. This model was used to evaluate whether suppressing gemcitabine-induced autophagy and the subsequent increase in collagen I in CAFs could restore the antitumor efficacy of gemcitabine. The coculture groups were treated as follows: the upper chambers were administered gemcitabine alone, gemcitabine plus CQ, or gemcitabine plus SC79. Except for the blank control group in the lower chamber, all other groups received gemcitabine. Cell viability in the lower chamber was measured using CCK8 assay to assess the impact of gemcitabine-induced CAF autophagy on its antitumor activity (Supplementary Fig. S2A).
CCK-8 shows that, in the Transwell coculture system, when CAFs in the upper chamber are treated with gemcitabine (Group 3), the sensitivity of pancreatic cancer cells in the lower chamber to gemcitabine is significantly decreased, and cell viability is markedly higher than that in Group 2 (upper chamber without CAFs). These findings indicate that the antitumor effect of gemcitabine is attenuated. In contrast, when the autophagy inhibitor CQ or the AKT activator SC79 is added to CAFs in the upper chamber to inhibit gemcitabine-induced autophagy (Groups 4 and 5), the antitumor activity of gemcitabine is largely restored, and cell viability in the lower chamber is reduced by approximately 20% compared with Group 3. These results demonstrate that gemcitabine-induced autophagy in CAFs desensitizes pancreatic cancer cells to gemcitabine, whereas inhibition of such CAF autophagy effectively enhances the sensitivity of pancreatic cancer cells to gemcitabine.
Next, we divided CAFs into four groups and used enzyme-linked immunosorbent assay (ELISA) to measure the expression level of collagen I precursor in the CAF supernatant, reflecting changes in collagen I secretion in the coculture system (Supplementary Fig. S2B). The results show that gemcitabine administration significantly increases collagen I precursor in the CAF supernatant, whereas combined treatment with CQ or SC79 to inhibit CAF autophagy markedly reduces its level. Together with the CCK-8 results from the coculture system, these findings further confirm that inhibiting CAF autophagy reduces collagen I secretion and thereby restores the sensitivity of tumor cells to gemcitabine.
To furthermore investigate whether combining CQ enhances the sensitivity of CAFs to gemcitabine, we performed CCK8 assays to measure cell viability and found cotreatment decreased cell viability compared with gemcitabine alone (Fig. 5A). Furthermore, treatment with CQ alters the apoptotic profile of gemcitabine-treated cells. Compared with the gemcitabine-alone group, the combination group exhibits decreased expression of the antiapoptotic protein Bcl-2 and increased expression of pro-apoptotic Bax, indicating enhanced apoptosis (Fig. 5B).
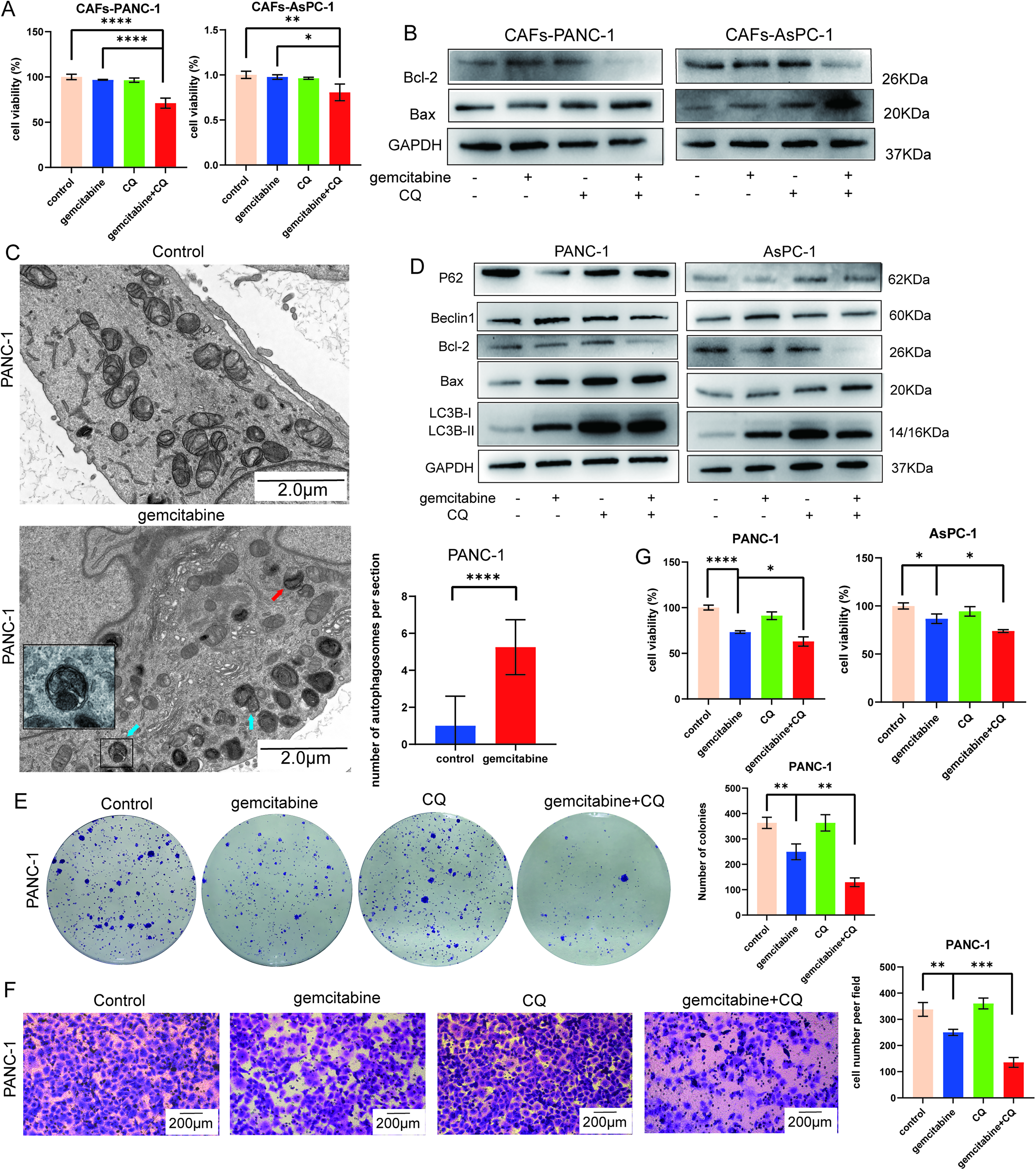
Combination with CQ enhances the cytotoxicity of gemcitabine against CAFs and pancreatic cancer cells.
Meanwhile, we also investigated the effects of combination therapy in pancreatic cancer cell lines. TEM was used to observe autophagosomes (Fig. 5C), and western blotting was performed to validate changes in autophagic and apoptotic proteins. Gemcitabine similarly induces autophagy in pancreatic cancer cells, as evidenced by increased levels of Beclin1 and LC3BII and decreased p62 compared with the control group (Fig. 5D). Given that gemcitabine can also induce autophagy in pancreatic cancer cells and tumor cells can also produce collagen I, we compared the expression levels of COL1A1 in pancreatic cancer cells and CAFs using reverse transcription quantitative polymerase chain reaction (RT-qPCR) and WB. The results show that COL1A1 mRNA and protein levels in tumor cells are low regardless of treatment with gemcitabine alone or combined with CQ to inhibit autophagy, compared with those in CAFs (Supplementary Fig. S3). These findings further confirm that the gemcitabine-induced increase in collagen I is mainly derived from CAFs rather than tumor cells. Furthermore, cotreatment with CQ inhibits autophagy and enhances apoptosis in pancreatic cancer cells (Fig. 5D). In addition, colony formation, Transwell, and CCK8 assays (Fig. 5E–G) demonstrate that cotreatment with CQ enhances the sensitivity of pancreatic cancer cells to gemcitabine, leading to further suppression of cell proliferation, migration, and survival compared with gemcitabine alone.
Combination of gemcitabine and CQ inhibits tumor growth and reduces collagen I in mouse model
To investigate the in vivo efficacy of CQ combined with gemcitabine, we subcutaneously injected murine pancreatic cancer cells PAN02 and mouse adipose-derived stem cells into C57 mouse. Mice were grouped and treated for 2 weeks, with tumor size monitored until 1 week after treatment cessation. Tumors were harvested at the end of the experiment (Fig. 6A). The tumor growth curve shows that the combination group exhibited the slowest tumor growth (Fig. 6B) and the fewest liver metastases (Fig. 6C). In addition, a higher number of TUNEL-positive cells is observed in the combination group (Fig. 6D), suggesting increased cell apoptosis.
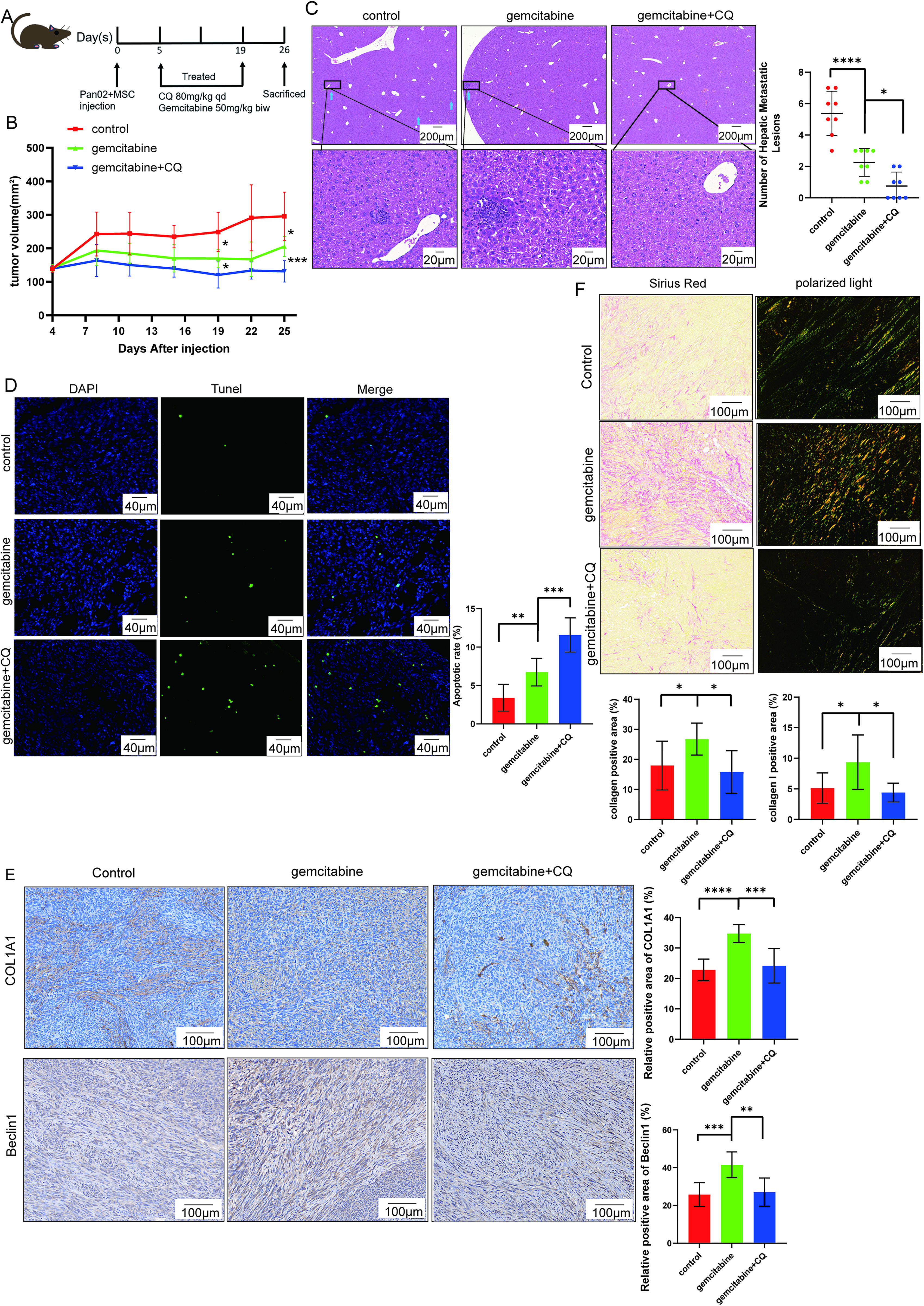
Combination of gemcitabine and CQ in mouse model (n = 8 per group).
To validate our in vitro findings, we detected P62, Beclin1, and COL1A1 expression in mouse tumor tissues. Compared with the control group, gemcitabine treatment downregulated P62 expression and upregulated Beclin1 and COL1A1 levels, whereas this effect was reversed by combination with CQ (Fig. 6E and Supplementary Fig. S4). Meanwhile, double immunofluorescence staining shows that apoptosis and autophagy are present in both tumor cells and CAFs (Supplementary Fig. S5). Sirius red staining further shows that gemcitabine significantly increases collagen deposition, as evidenced by prominent orange-yellow collagen I under polarized light microscopy, which is markedly attenuated by CQ cotreatment (Fig. 6F).
Finally, to further clarify the relationship between autophagy in CAFs and collagen I deposition, we performed multiplex immunofluorescence staining on mouse tumor tissues from the three groups, co-staining for COL1A1, BECN1, and α-SMA (Supplementary Fig. S6). Compared with the control group and the combined group, the gemcitabine-treated group shows distinct punctate Beclin1 (yellow) accumulation in the cytoplasm of α-SMA-positive (green) CAFs, indicating that gemcitabine activates autophagy in CAFs. Meanwhile, COL1A1-positive (red) regions are observed in close proximity to these autophagy-active CAFs, supporting a correlation between CAF autophagy and collagen I. Further quantitative analysis reveals that both CAF autophagy and COL1A1 levels are elevated in the gemcitabine group relative than in the control group, and this gemcitabine-induced increase is reversed by cotreatment with the CQ. These results indicate that gemcitabine-induced collagen I deposition is associated with the activation of autophagy in CAFs.
Discussion
Our study explained how gemcitabine induces collagen accumulation from the perspective of CAF autophagy, revealing that gemcitabine decreased AKT phosphorylation to trigger CAF autophagy, which in turn drove aberrant deposition of collagen I. In conjunction with our clinical observations, increased collagen expression after gemcitabine treatment in patient specimens, together with the positive correlation between COL1A1 and gemcitabine resistance from CTRP, supports a biologically plausible inference that gemcitabine-induced collagen deposition may be associated with reduced sensitivity to gemcitabine. Our subsequent experiments also confirm that inhibiting CAF autophagy could reduce collagen accumulation and enhance the antitumor effect of gemcitabine. In addition, our findings suggest that COL1A1 may serve as a biomarker to stratify patients who would benefit most from combination therapy, offering a rationale and potential biomarker for the application of hydroxychloroquine (HCQ) in pancreatic cancer patients, especially in neoadjuvant and adjuvant stages.
We found that combining gemcitabine with autophagy inhibitor CQ suppressed autophagy and reduced collagen I expression, and these findings are consistent with existing literature. Bai et al. presented inhibited autophagy in CAFs, particularly mitophagy, impeding collagen I synthesis. Mechanistically, autophagy promotes proline production by regulating mitochondrial NAD kinase 2, which serves as an essential precursor for collagen synthesis. 13 These observations imply that gemcitabine may indirectly regulate proline metabolism and collagen production in CAFs, possibly through the induction of autophagy. Further investigation is warranted to elucidate this process. Yan et al. found Yes-associated protein 1 (YAP)+CAFs could activate autophagy and upregulate COL1A1 expression. 28 In hypertrophic scar models, inhibiting autophagy effectively suppressed fibrotic progression and reduced the expression of collagen I. 29 Meanwhile, a few studies have suggested a role for autophagy in collagen degradation. Liu et al. found that leucine promoted collagen deposition by inhibiting autophagy in a fish model. 30 Zhao et al. also proposed that impaired mitophagy in renal mesangial cells led to extracellular matrix deposition and promoted renal fibrosis. 31 Of note, these studies were conducted in nontumor models, and the cells used were neither tumor cells nor stromal cells. Furthermore, Hu et al. reported that high expression of autophagy-related proteins in renal tubular cells promoted fibrosis. 32 Therefore, the relationship between autophagy and collagen deposition may vary depending on different models and stimuli.
Gemcitabine-induced collagen I deposition may result from both reactive fibrosis and CAF-specific stress response. On the one hand, chemotherapeutic agents can cause tissue damage and trigger reactive fibrosis associated with tissue repair. 33 On the other hand, our study revealed that gemcitabine-induced collagen I accumulation is at least partly mediated by CAF autophagy, which is linked to downregulated AKT phosphorylation. The observed activation of autophagy and reduced AKT phosphorylation in CAFs confirm the presence of CAF-specific stress response, rather than mere tissue injury repair. However, the precise contribution of each mechanism cannot be clearly distinguished at present. In this study, gemcitabine upregulated COL1A1 protein and mRNA level by activating autophagy, suggesting that it promotes collagen I synthesis in CAFs. In addition, measurements of collagen I precursors in CAFs culture supernatants show that gemcitabine-induced autophagy also increases extracellular collagen I precursor secretion. We therefore propose that gemcitabine-induced autophagy in CAFs enhances both the synthesis and secretion of collagen I, consistent with increased collagen deposition observed by Sirius Red staining and IHC. However, our study cannot distinguish whether the observed collagen accumulation is driven primarily by altered synthesis or secretion. This process likely involves both cytoprotective and secretory autophagy, which may act together to promote collagen I deposition following gemcitabine treatment.
Our findings indicate that, compared with autophagy inhibitors, the AKT agonist only partially reverses gemcitabine-induced upregulation of COL1A1. This suggests that the regulation of autophagy and collagen I in CAFs involves additional signaling pathways beyond AKT during gemcitabine-induced autophagy and collagen accumulation. Previous studies have demonstrated that activation of the transforming growth factor-β/mothers against decapentaplegic homolog 2 (TGF-β/Smad2) pathway induces autophagy and promotes the differentiation of fibroblasts into myofibroblasts in oral mucosal fibrosis models. 34 Moreover, TGF-β/Smad2 signaling stimulates the expression of extracellular matrix components, including collagen, and inhibition of this pathway reduces extracellular matrix deposition, such as collagen, thereby enhancing the therapeutic efficacy of gemcitabine. 35 Therefore, gemcitabine-mediated autophagy and subsequent collagen deposition in CAFs may rely on other pathways in addition to AKT signaling, such as the TGF-β/Smad2 axis. Further investigation is warranted to better target collagen deposition and improve therapeutic outcomes.
In general, as a nucleoside analog, gemcitabine can induce DNA damage by blocking DNA synthesis and repair, ultimately leading to apoptosis. 36 During this process, cells consume large amounts of ATP,37–39 which may indirectly affect the phosphorylation of key proteins in multiple signaling pathways, resulting in decreased phosphorylation levels.40,41 In contrast to these studies, our results show that gemcitabine did not cause obvious cell death in CAFs, suggesting that the mechanism by which gemcitabine downregulates AKT phosphorylation in CAFs is unlikely to be mediated by the induction of apoptosis. Studies have shown that chemotherapeutic drugs can induce metabolic reprogramming. 42 As a nucleoside analog, gemcitabine affects nucleotide metabolism. Liu et al. reported that gemcitabine can induce dual activation of the adenosine monophosphate (AMP)-cAMP axis, further activating the downstream AMP-activated protein kinase, 43 whose activation negatively regulates the AKT pathway. This suggests that gemcitabine may downregulate AKT phosphorylation indirectly by affecting nucleotide metabolism, triggering metabolic reprogramming and adaptive stress in CAFs. Therefore, although a potential role of DNA damage-related signaling cannot be completely ruled out, the reduction in AKT phosphorylation in CAFs induced by gemcitabine observed in our study is more likely a consequence of adaptive metabolic stress in CAFs.
However, some studies have suggested that complete stromal or collagen depletion may promote pancreatic cancer progression. Chen et al. found that complete ablation of collagen I in α-SMA+ myofibroblasts in mouse models conversely promoted immunosuppression, accelerated pancreatic cancer progression, and reduced OS in mice. 44 Nevertheless, the regulation of tumor stroma is highly complex. In addition to collagen, the stroma contains various components, including hyaluronan and fibronectin. 45 A previous study demonstrated that autophagy can degrade uridine diphosphate (UDP)-glucose dehydrogenase (UGDH), thereby inhibiting hyaluronan synthesis, whereas inhibition of autophagy leads to UGDH accumulation and increased hyaluronan production. 46 This suggests that inhibiting autophagy in CAFs may not result in complete stromal depletion. Therefore, in the targeting of tumor stroma, consideration should be given to both improving the physical barrier to enhance drug delivery and avoiding tumor progression caused by complete stromal depletion.
Currently, there are clinical studies investigating the use of gemcitabine combined with HCQ in pancreatic cancer patients. A phase II study of AG combined with HCQ shows that adding HCQ improves the overall response rate in patients with metastatic pancreatic cancer, suggesting that locally advanced patients may derive greater benefit from combination therapy. 47 In addition, a phase II neoadjuvant clinical study in pancreatic cancer finds combining HCQ increases the pathological response rate. 48 However, no large-scale clinical studies have yet demonstrated that combination therapy improves OS. This suggests a need to identify biomarkers and select patients at appropriate disease stages. Furthermore, in current clinical studies on adding HCQ to neoadjuvant therapy, HCQ was not included in the subsequent adjuvant therapy, which may also contribute to the lack of OS benefit.
We hypothesize that gemcitabine combined with HCQ may increase the pathological response rate by inhibiting CAF autophagy, reducing collagen deposition, remodeling the tumor stroma, and enhancing chemotherapeutic drug delivery. As a predictive marker for surgical resection rate and local control, pathological response carries greater clinical significance than OS in resectable or borderline resectable pancreatic cancer. In addition, studies suggest that the neoadjuvant phase represents the optimal therapeutic window for inhibiting CAF activation, during which the collagen network has not yet solidified; regulating its subtypes can significantly improve therapeutic efficacy.49,50 We hypothesize that the HCQ combination regimen can reduce collagen production and may create favorable conditions for surgical resection. Meanwhile, studies have shown that the stroma in metastatic patients may already become fibrotic,51,52 limiting the benefit of stromal intervention. Therefore, integrating our study with previous clinical research, neoadjuvant or adjuvant therapy may be more suitable for combination therapy, and priority could be given to patients with high COL1A1 expression.
Tumor cells also produce a small amount of collagen I. Chen et al. reported that pancreatic cancer cells can synthesize a unique homotrimeric collagen I, which is distinct from the classical heterotrimeric collagen produced by fibroblasts in normal tissues. Notably, the collagen homotrimer synthesized by pancreatic cancer cells only contains the α1 chain. Inhibition of this collagen I homotrimer has been shown to enhance T-cell infiltration and improve the efficacy of immunotherapy. 53 Although the present study did not further explore the effect of gemcitabine-induced autophagy in tumor cells on collagen deposition, the above findings clearly indicate that the contribution of tumor cells to collagen production and remodeling cannot be overlooked. This issue will be further investigated in our future research.
However, our study also has limitations. In this study, all clinical specimens were FFPE tissues, and paired fresh biopsy and surgical samples from the same patients were unavailable. Therefore, we were unable to verify collagen expression by western blotting or other quantitative methods. Future studies with fresh clinical samples and more quantitative detection approaches are warranted to further confirm our findings. Meanwhile, verifying the present findings using primary CAFs isolated from human pancreatic cancer tissues will be of greater significance in future research and will also enhance the clinical translational value of this study. As for animal models, survival analysis was not performed in this study and will be supplemented in future investigations to enhance clinical relevance. Finally, the clinical cohort is small (n = 8) with substantial inter-patient heterogeneity, which limited the statistical power of correlation analysis between CAF autophagy and collagen deposition. Future studies with larger cohorts will help validate these clinical observations.
Conclusion
Gemcitabine increases collagen I in CAFs in association with downregulation of the AKT/autophagy axis in CAFs, impairing its own efficacy (Fig. 7). Future small-scale clinical studies could first be conducted in patients with high COL1A1 expression to verify the efficacy of gemcitabine combined with autophagy inhibitor in neoadjuvant or adjuvant therapy for pancreatic cancer.
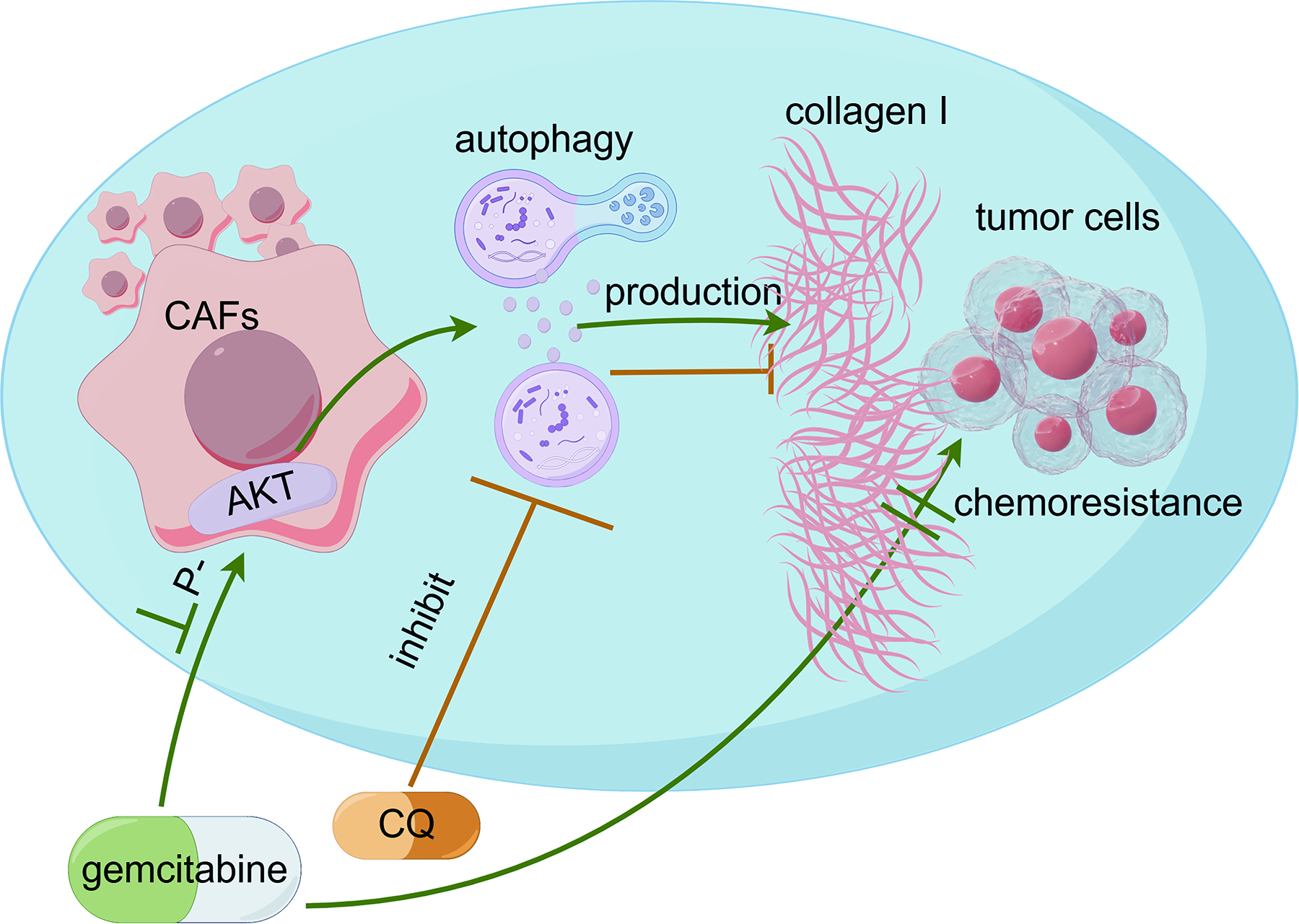
Proposed mechanism of gemcitabine-induced fibrosis and the enhancing effect of CQ on chemotherapeutic efficacy. Gemcitabine downregulates AKT phosphorylation in CAFs, thereby triggering autophagy. This induction of autophagy promotes the aberrant accumulation of collagen I, forming a physical barrier that impairs chemotherapeutic efficacy. Cotreatment with CQ blocks autophagic flux, reduces collagen deposition, and enhances the therapeutic effect of gemcitabine.
Footnotes
Acknowledgments
The schematic diagrams were created with the support of Figdraw.
Ethics Statement
This study involving experimental animals was conducted in strict accordance with the Regulations on the Administration of Experimental Animals of the People’s Republic of China.
Author Disclosure Statement
All authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.
Funding Information
This study was supported by the
Supplemental Material
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.