
Abstract
The cancer stem cell (CSC) paradigm has evolved from a rigid hierarchical model to a systems-level perspective in which stemness is a reversible and context-dependent phenotype. Evidence from lineage tracing and single-cell/spatial multiomics indicates that tumor cells occupy continuously evolving phenotypic states governed by complex gene regulatory networks. Within this landscape, CSCs can be interpreted as metastable attractors maintained through coupled signaling, epigenetic, metabolic, transcriptional, and microenvironmental interactions. Tumor heterogeneity and therapeutic resistance emerge through phenotypic reprogramming, regulatory network rewiring, and niche-dependent stabilization under environmental and therapeutic stress. This reframes resistance as an emergent property of tumor ecosystems, underscoring the limitations of targeting static CSC populations or single pathways. Therefore, durable therapeutic control will require network-oriented interventions capable of reshaping attractor topology and disrupting stemness-supportive microenvironments.
Keywords
Introduction
The cancer stem cell (CSC) hypothesis was originally proposed as a hierarchical model in which tumors are organized analogously to normal tissues, with a rare subpopulation of stem-like cells responsible for self-renewal, differentiation, and long-term tumor maintenance. This framework was first experimentally validated in acute myeloid leukemia, where only a small fraction of leukemic cells exhibited tumor-initiating capacity in xenotransplantation assays. 1 This early hierarchical interpretation of CSCs aligns with broader advances in stem cell biology and regenerative medicine, where translational stem cell research has demonstrated the clinical and experimental feasibility of manipulating stem-like populations in disease contexts. 2 The hypothesis was subsequently extended to solid tumors, including breast cancer, where phenotypically defined subpopulations demonstrated enhanced tumorigenicity and self-renewal potential. 3
Advances in lineage tracing, single-cell RNA sequencing, spatial transcriptomics, and multiomics profiling have substantially revised this hierarchical paradigm. Evidence indicates that tumor-propagating potential is not restricted to a stable CSC subset. Nonstem cancer cells can regain stem-like features and reconstitute tumor heterogeneity following the depletion of CSC-enriched populations, demonstrating bidirectional and reversible state transitions.4,5 Single-cell and spatial analyses further show that tumors consist of interconverting cellular populations distributed along phenotypic continua rather than discrete hierarchical compartments.6,7
This plasticity-centered perspective aligns with Waddington’s epigenetic landscape framework, in which cellular identity reflects movement across multidimensional regulatory terrains rather than irreversible differentiation trajectories. In tumors, genetic alterations, epigenetic remodeling, and microenvironmental signals reshape these landscapes, lowering transition barriers between phenotypic states and enabling adaptive reprogramming under stress.8,9 Additionally, the tumor microenvironment (TME) acts as an active regulatory system that shapes tumor cell identity through biochemical, mechanical, and immune-mediated signaling. Developmental pathways such as Wnt/β-catenin, Notch, and TGF-β integrate niche-derived cues with intracellular regulatory circuits to stabilize stem-like phenotypes and promote adaptive responses.10,11
Systems biology conceptualizes CSCs as metastable attractor states embedded within interconnected regulatory networks. In this framework, cellular phenotypes emerge from nonlinear interactions among transcriptional circuits, signaling pathways, epigenetic regulators, and metabolic networks that collectively define stable and metastable gene expression states.12,13 Single-cell and spatial omics studies further demonstrate that tumor cells occupy structured yet dynamic state spaces, where transitions between attractor states can be induced by intrinsic stochasticity or extrinsic perturbations, including therapeutic stress and immune pressure. 14
This review synthesizes experimental oncology, single-cell biology, cancer systems biology, and dynamical systems theory to present CSCs as metastable, ecosystem-based regulators of tumor heterogeneity and therapy resistance. Uniquely, it integrates multiomics data with dynamical systems theory to redefine stemness as a reversible ecosystem property and to conceptualize CSCs as controllable attractor states for network-based therapeutic reprogramming.
Historical Evolution of the CSC Concept: From Hierarchy to Plasticity
The classical hierarchical model, supported by xenotransplantation assays, proposed that tumors are organized analogously to normal tissues, with rare CSCs at the apex. However, these assays are affected by cross-species differences in stromal interactions and immune responses, which often lead to an underestimation of tumorigenic potential. In melanoma models under optimized conditions, a significantly larger fraction of cells can initiate tumors, challenging the notion that CSCs are rare and fixed. 5 Figure 1 provides a schematic representation of the shift from a unidirectional hierarchy to a dynamic attractor landscape.
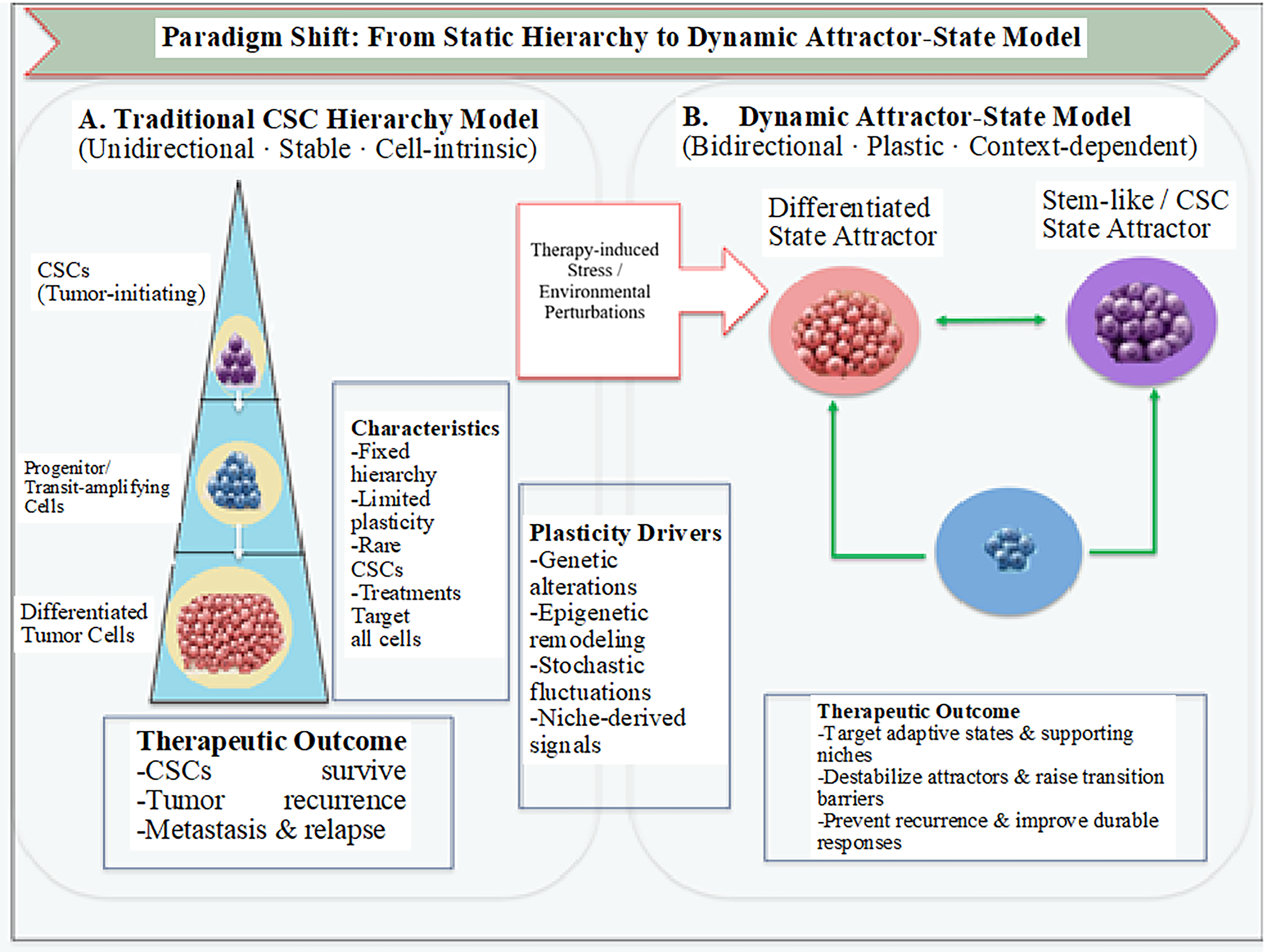
Transition from CSC hierarchy to dynamic attractor-state model. CSC, cancer stem cell.
More definitive evidence against a strictly unidirectional hierarchy has emerged from in vivo lineage tracing, demonstrating that tumor-propagating capacity is not confined to a rare, stable CSC pool. Differentiated tumor cells can reacquire stem-like properties through dedifferentiation, for example, in intestinal tumor models, ablation of Lgr5+ CSCs led to repopulation by nonstem cells. 4 This capacity for dedifferentiation marked a major conceptual turning point, establishing self-renewal capacity as a fluid, environmentally regulated phenotype driven by epigenetic remodeling, transcriptional reprogramming, and metabolic adaptation. 14
Therapeutic stress strongly promotes phenotypic state transitions. Exposure to cytotoxic agents induces transient drug-tolerant states characterized by enhanced survival and activation of stemness-associated programs. These persister populations can reenter proliferative programs following treatment withdrawal. 15 Large-scale single-cell analyses further demonstrate widespread phenotypic variability and adaptive reprogramming across tumor types, with cells continuously shifting along a spectrum of phenotypic states. 16 Transcriptional noise and stochastic gene-expression variability can spontaneously shift cells between phenotypic configurations, even within genetically identical populations. 17
The Attractor-State Framework: CSCs as Dynamic Systems-Level Entities
Recent advances have challenged the traditional view of CSCs as a fixed, rare subpopulation. Instead, CSCs are increasingly recognized as plastic, niche-dependent phenotypic states that emerge from complex regulatory networks. Table 1 summarizes this conceptual shift, while Figure 2 illustrates how therapeutic stress drives cells across a regulatory barrier into a stem-like attractor state.
Evolution of the CSC Concept from Hierarchical Organization to Dynamic Attractor-State Systems

CSC, cancer stem cell; GRN, gene regulatory network.
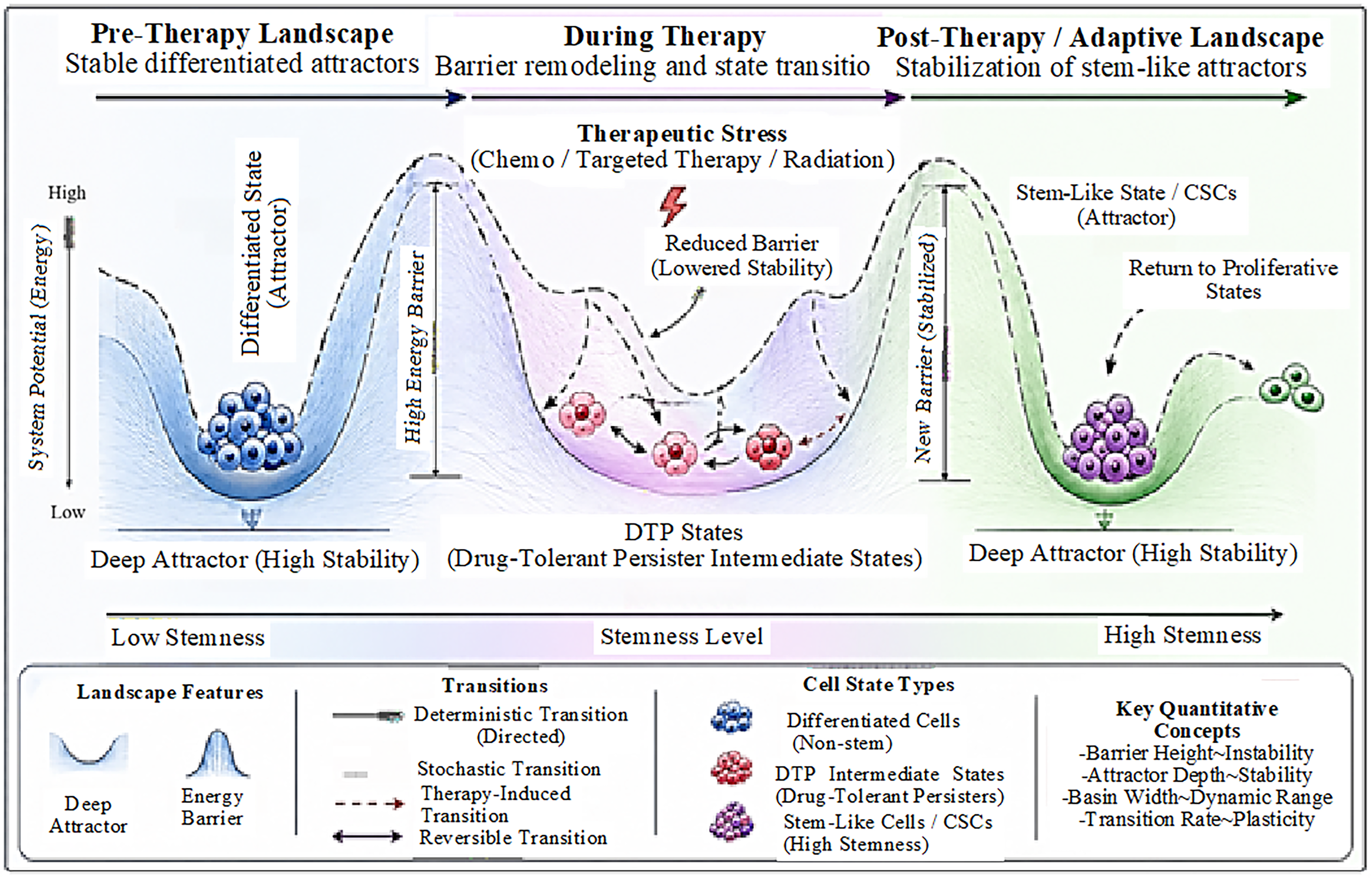
Therapy-induced transitions across a high-dimensional landscape illustrating destabilization of differentiated states and emergence of drug-tolerant stem-like attractors under therapeutic stress.
Foundations of dynamical systems theory in cancer biology
In dynamical systems theory, attractors are stable regions of regulatory state space toward which cellular trajectories converge despite stochastic molecular fluctuations. CSC attractors, therefore, represent robust yet reversible configurations of coordinated transcriptional and signaling activity. Cellular phenotypes correspond to stable or metastable attractor states arising from coordinated gene expression patterns. Single-cell transcriptomic analyses support this perspective, demonstrating that tumor cells occupy structured, continuous state spaces shaped by regulatory interactions. 16 A defining feature of attractor states is robustness to molecular noise while remaining responsive to perturbations that trigger phenotypic transitions into alternative cellular configurations. 15
Time-resolved single-cell analyses demonstrate that cancer cells explore dynamic regions of the regulatory landscape, occasionally transitioning between attractors. 14 Lineage-tracing studies functionally validate that tumor cells transition bidirectionally between stem-like and differentiated states in response to microenvironmental cues or pharmacological perturbations. 4
Importantly, attractor landscapes remain dynamically remodelable in cancer. Perturbations such as genetic mutations, microenvironmental cues, and therapy-induced stress can reshape the topology of this landscape, altering the number, stability, and accessibility of phenotypic states.12,17 These observations support a unified, integrative view in which CSCs are dynamic, condition-specific states that emerge from the interplay between regulatory network architecture and environmental perturbations.
Stemness as an emergent systems-level property
Within the attractor-state framework, stem-like behavior emerges from coordinated interactions among transcriptional, epigenetic, signaling, and metabolic programs. High-resolution single-cell transcriptomic studies show that stem-like phenotypes are distributed along continuous gene expression gradients rather than confined to discrete subpopulations.6,8 Core pluripotency regulators including OCT4 and SOX2 participate in autoregulatory transcriptional circuits associated with stemness maintenance. 24 Microenvironmental regulation of stem cell senescence further demonstrates that stem-like states are not fixed but are dynamically maintained through extrinsic niche conditions, which modulate cellular aging and functional plasticity. 25 Perturbation studies indicate that disrupting system-wide connectivity destabilizes stem-like phenotypes more effectively than targeting isolated molecular components. 19
Transcriptional circuits are closely integrated with extracellular signaling pathways. Mechanotransduction mediated by YAP/TAZ links extracellular matrix (ECM) stiffness to transcriptional programs that stabilize stem-like phenotypes, modulating chromatin accessibility and maintaining stemness.26,27 Epigenetic remodeling further regulates stemness by altering chromatin accessibility and facilitating transitions between phenotypic states.22,23 Pan-cancer studies have developed quantitative stemness indices based on transcriptomic and epigenomic features, demonstrating that stem-like characteristics correlate with coordinated network-level signatures.24,28
Therapy-induced plasticity and resistance as dynamic state transitions
Within the attractor-state framework, therapeutic resistance is increasingly understood as a systems-level adaptive process rather than the expansion of a fixed resistant clone. Therapeutic stress reshapes tumor regulatory landscapes through coordinated alterations in signaling activity, chromatin accessibility, metabolic programs, and microenvironmental interactions, thereby destabilizing differentiated attractor states and promoting transitions into stem-like or drug-tolerant phenotypes.17,20 Lineage-tracing and single-cell studies further demonstrate that non-CSCs can reversibly reacquire stemness-associated properties during treatment, replenishing tumor-propagating populations and restoring tumor heterogeneity following therapy withdrawal. 4
These adaptive transitions frequently involve quiescent or slow-cycling DTP states characterized by enhanced survival capacity, stress tolerance, and resistance to apoptosis.29,30 Longitudinal single-cell and multiomic analyses further indicate that resistant phenotypes frequently emerge during treatment through coordinated transcriptional and epigenetic reprogramming rather than solely through preexisting genetic mutations.15,31
Microenvironmental signals further reinforce these transitions by lowering barriers between phenotypic states. Hypoxia, inflammatory cytokines, stromal remodeling, and metabolic stress collectively stabilize dedifferentiated attractors and sustain phenotypic flexibility under therapeutic pressure.9,21 Consequently, resistance emerges as an ecosystem-level property shaped by reciprocal interactions among tumor cells, stem-like populations, and microenvironmental compartments.9,31 This system-level perspective helps explain the limited durability of single-pathway therapies and highlights the importance of strategies capable of destabilizing adaptive attractor states together with their supporting microenvironments.
Regulatory Networks Governing CSC Attractor States
CSC phenotypes are maintained through interconnected signaling and transcriptional circuits that integrate biomechanical, inflammatory, metabolic, and microenvironmental inputs. 11 Developmental and stress-responsive pathways, including Wnt/β-catenin, Notch, Hedgehog, JAK/STAT, NF-κB, TGF-β, and Hippo–YAP/TAZ pathways, form interconnected regulatory modules with extensive feedback and compensatory crosstalk. Genome-scale CRISPR perturbation studies show that inhibition of a single pathway is often insufficient to abolish stem-like phenotypes due to compensatory activation of parallel signaling circuits that preserve CSC functionality. 32 Table 2 illustrates the integrated signaling networks that stabilize CSC attractor states.
Integrated Regulatory Network Modules Governing CSC Attractor-State Stability and Plasticity

ECM, extracellular matrix.
Mechanical and inflammatory microenvironmental inputs play major roles in reinforcing CSC attractor states. Wnt/β-catenin and Hippo–YAP/TAZ pathways act as mechanotransduction hubs, where ECM stiffness and cytoskeletal tension activate YAP/TAZ to cooperate with β-catenin in driving stemness-associated transcriptional programs and tumor initiation.27,41 In parallel, inflammatory signaling reinforces CSC maintenance through coordinated Notch–NF-κB and IL-6/JAK/STAT3 modules. NF-κB-driven cytokine signaling enhances Notch activation, while reciprocal feedback sustains inflammatory niches that support CSC persistence.33,34 IL-6/JAK/STAT3 signaling further promotes CSC survival, proliferation, EMT-like transitions, and therapy-adaptive states under inflammatory stress.35,36
TGF-β signaling integrates EMT programs with epigenetic and transcriptional regulation to enable reversible dedifferentiation and phenotypic plasticity. EMT transcription factors such as Snail and ZEB1 repress epithelial identity while activating stemness-associated gene programs through chromatin remodeling. 37 These transitions are stabilized through a bistable regulatory circuit, particularly the miR-200/ZEB axis, which enables reversible transitions between epithelial and mesenchymal stem-like states in response to hypoxia, inflammatory signals, and TGF-β signaling. 39 MicroRNA-mediated regulation, together with bidirectional communication between CSCs and stromal and immune cells, further reshapes the regulatory landscape and enhances phenotypic adaptability. 37
The redundancy, feedback loops, and compensatory interactions within these networks confer strong robustness to CSC attractor states. As a result, pathway inhibition often triggers adaptive network rewiring rather than system collapse, enabling CSCs to maintain functional stability under therapeutic pressure. 42 Collectively, these pathways form resilient signaling networks linked through shared transcriptional outputs and niche-derived feedback mechanisms. 43 Consequently, combinatorial therapeutic strategies targeting multiple nodes within this network architecture, such as dual Wnt/YAP/TAZ inhibition, coordinated Notch/NF-κB blockade, or combined JAK/STAT inhibition with epigenetic modulators, have demonstrated greater efficacy in destabilizing CSC states and reducing tumor-initiating capacity than single-pathway approaches. 40
The CSC Niche: Co-Evolution Beyond Passive Support
Various components, including mechanical modulators, metabolic conditions, immune interactions, and neural signaling, collectively shape and sustain CSC phenotypes. Table 3 outlines the key components of the CSC niche, while Figure 3 schematically illustrates the integration of these mechanical, metabolic, immune, and neural elements within this complex ecosystem.
Multiscale Components of the CSC Niche and Their Roles in Attractor-State Stabilization

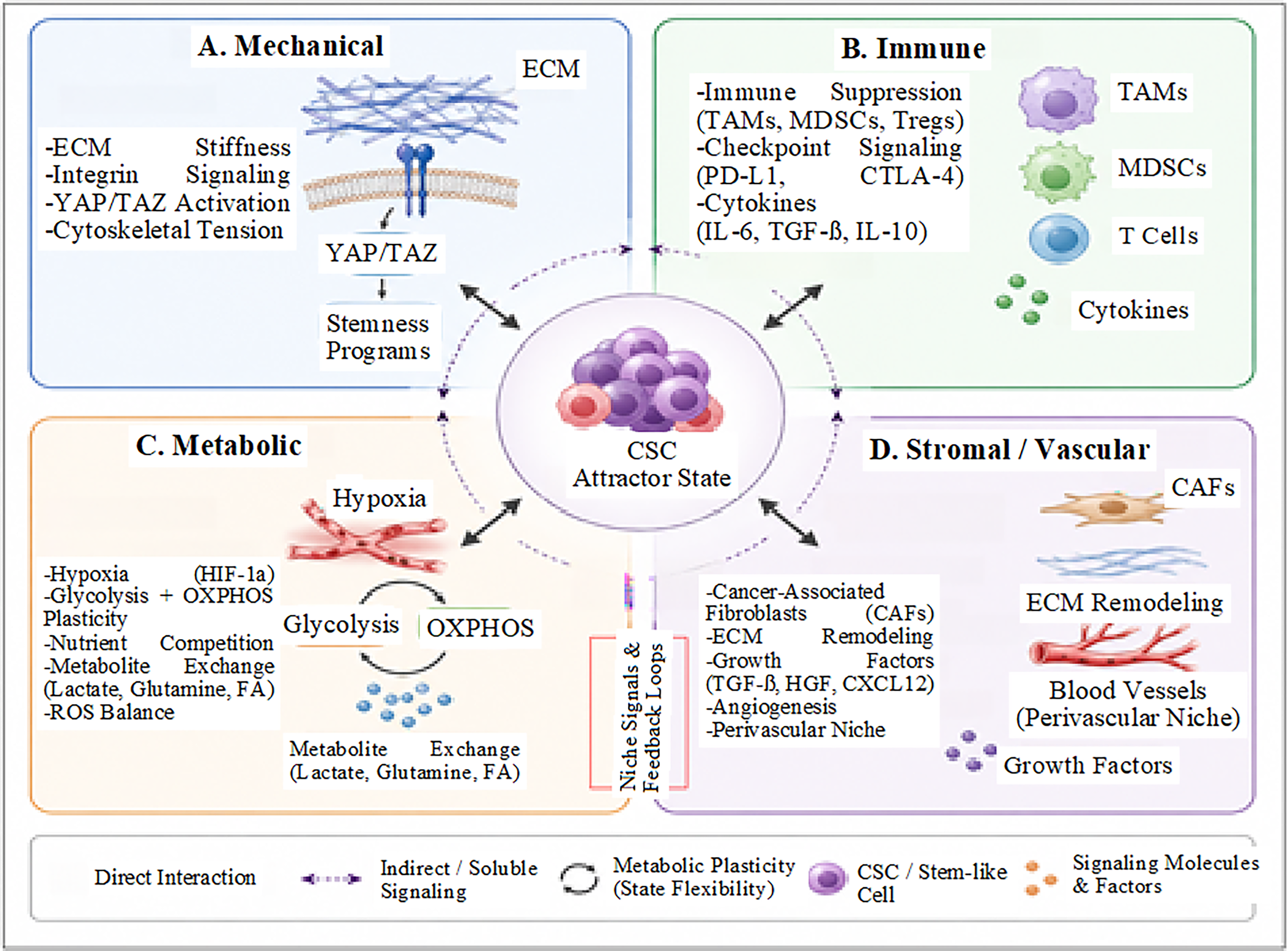
Multiscale tumor microenvironment as an attractor-stabilizing ecosystem.
The niche as a dynamic, co-evolving ecosystem
The CSC niche functions as a continuously remodeling microenvironmental system in which tumors, stromal, immune, and vascular compartments exchange biochemical and spatial information. Spatial and lineage-tracing studies show that CSCs actively participate in niche construction by secreting cytokines, growth factors, and extracellular vesicles (EVs) that reprogram surrounding cells into tumor-supportive states. 53 Stromal-stem cell interactions, particularly involving adipose-derived stromal cells, have been shown to modulate tumor progression in a cancer subtype- and age-dependent manner, highlighting the dynamic reciprocity between tumor cells and their microenvironment. 54 High-resolution spatial profiling further demonstrates that CSC-enriched regions are defined by coordinated signaling, metabolic adaptation, and immune regulation, indicating that stemness emerges from integrated microenvironmental constraints rather than cell-intrinsic programs alone.44,46 Overall, the niche stabilizes CSC phenotypes through reciprocal signaling between tumor cells and surrounding microenvironmental compartments.
Mechanical and metabolic niches
Mechanical properties of the TME regulate CSC states by transducing extracellular forces into transcriptional programs, primarily through integrin–FAK–Src signaling converging on YAP/TAZ-dependent stemness networks. 55 Rather than structural remodeling per se, ECM mechanics function as dynamic inputs that stabilize stem-like transcriptional states and coordinate tumor progression within evolving microenvironmental contexts. 56
In parallel, CSCs exhibit metabolically adaptive phenotypic plasticity, enabling transitions between glycolytic and oxidative states depending on nutrient and oxygen availability. This metabolic adaptability supports survival under environmental and therapeutic stress by maintaining energy balance and redox homeostasis. 48 Single-cell metabolic analyses further confirm that CSC metabolic states are not fixed but dynamically reprogrammed in response to spatial gradients within tumors, reinforcing therapy resistance and persistence.49,57
Therapeutic Resistance as an Emergent Property
Therapeutic resistance emerges from coordinated interactions among tumor cells, CSC-like populations, and microenvironmental compartments rather than purely cell-autonomous genetic mechanisms. Single-cell studies demonstrate that therapy reorganizes tumor populations through adaptive transcriptional and epigenetic reprogramming. At the systems level, resistance reflects dynamic state reconfiguration within a coupled regulatory and ecological network, rather than a strict genetic phenomenon.
Niche-integrated resistance mechanisms in CSCs
Therapeutic resistance in CSCs arises from a niche-dependent system integrating spatial protection, metabolic adaptation, and epigenetically mediated state transitions. The CSC niche forms protective stromal- and vascular-rich microdomains that restrict drug penetration while sustaining prosurvival signaling. 58 Hypoxia, aberrant vasculature, and ECM remodeling further reinforce stress-adapted phenotypes by stabilizing therapy-tolerant cellular states. 45
Within this microenvironment, CSCs exhibit metabolic plasticity, switching between glycolytic and oxidative phosphorylation programs in response to nutrient and oxygen stress. Therapy often selects for oxidative phosphorylation-biased states associated with enhanced redox control and survival under oxidative damage. 49 In parallel, metabolic exchange with stromal compartments, such as lactate shuttling and glutamine utilization, supports ecosystem-level adaptation under treatment pressure. 59
Epigenetic regulation provides a key mechanism for reversible nongenetic adaptation. Therapy induces chromatin remodeling that generates transcriptionally permissive but reversible states, enabling rapid reactivation of stemness-associated programs after treatment withdrawal.60,61 Together with DNA methylation and histone modifications, these changes maintain CSCs in a drug-tolerant, often quiescent state that facilitates relapse. This epigenetic flexibility represents a core driver of adaptive resistance by stabilizing transient survival attractors rather than fixed genetic resistance programs.
Immune evasion in CSC-driven resistance
CSCs employ coordinated immune evasion strategies that enhance survival under therapeutic and immunological pressure. Reduced MHC-I expression impairs antigen presentation, while PD-L1 upregulation suppresses T-cell activity. 50 Additional mechanisms, including antiphagocytic signaling and cytokine-mediated immunosuppression, reshape immune surveillance and reinforce tumor persistence.62,63
CSC-associated immune evasion is further reinforced by quiescence and stem-like states that reduce immunogenicity and increase resistance to immune-mediated clearance. These mechanisms operate within a coupled tumor–immune ecosystem in which immune pressure and tumor plasticity coevolve, producing persistent therapy-resistant states.28,29
Therapeutic Implications and Future Directions
Reframing therapeutic goals
Reconceptualizing CSCs as dynamic, systems-stabilized states shifts therapeutic focus from eradication of a fixed CSC population to disruption of the regulatory and microenvironmental interactions that sustain stemness. CSC identity is maintained through coupled signaling networks, metabolic programs, and niche-derived cues, making resistance an emergent property of tumor systems rather than a static cell trait. 7 Accordingly, durable therapeutic responses are more likely to arise from interventions that destabilize these interacting support systems rather than targeting CSC markers alone.
Mechanistically, modulation of tumor metabolism can reverse immunosuppressive and stemness-associated states, thereby enhancing tumor vulnerability. 64 Likewise, targeting biomechanical cues such as ECM stiffness can attenuate mechanotransduction-driven stemness programs and improve therapeutic accessibility of tumor regions. 65 Together, these findings suggest that durable CSC control will likely require simultaneous disruption of both intrinsic regulatory programs and supportive niche-derived signals.
Network-oriented and combinatorial approaches
The redundancy and feedback-rich structure of CSC regulatory networks limit the efficacy of single-pathway inhibition. Perturbation studies demonstrate that blockade of individual signaling routes is often compensated by alternative pathways, maintaining CSC survival and adaptability. 66 Consequently, combinatorial targeting of interconnected pathways provides greater suppression of CSC-like populations. For example, dual inhibition of Wnt and YAP/TAZ signaling more effectively reduces tumor initiation capacity than single-agent strategies, 40 while targeting cytokine-associated pathways such as IL-6/JAK/STAT and NF-κB disrupts inflammation-driven CSC maintenance. 36 Epigenetic therapies can further limit transcriptional flexibility required for state transitions under therapeutic stress. 67 Differentiation-based strategies also reduce tumorigenic potential by forcing CSCs into less plastic, terminal states. 68 Together, these findings support multitarget approaches that disrupt network robustness rather than isolated nodes.
Niche-destabilizing strategies
Disrupting the CSC niche is essential for overcoming microenvironment-mediated protection. Remodeling the ECM, including reduction of fibrosis and stiffness, improves drug penetration and weakens stemness-associated signaling. 69 Metabolic interventions that restrict mitochondrial function can further reduce CSC adaptability under stress conditions. In addition, combining metabolic or stromal targeting with immunotherapy enhances elimination of stem-like tumor populations. 70 Adaptive treatment strategies that modulate dosing over time can also suppress resistant subpopulations by preventing selective expansion of CSC states. 71
A complementary approach targets EV-mediated communication, which plays a central role in CSC-driven tumor progression. CSC-derived EVs contribute to immune evasion, metastatic niche formation, and therapy resistance by transferring bioactive cargo that reprograms recipient cells. 52 Accordingly, inhibition of EV release or uptake represents a promising strategy to disrupt intercellular signaling that sustains tumor evolution and metastatic competence. 72
Quantitative modeling of CSC attractor landscapes
Quantitative systems biology approaches increasingly support the reconstruction of CSC attractor landscapes and the prediction of phenotypic transitions associated with tumor plasticity and therapy resistance. Boolean and gene regulatory network models represent signaling and transcriptional circuits as interconnected regulatory nodes that generate stable attractors corresponding to stem-like, differentiated, or drug-tolerant states. These studies demonstrate that feedback interactions among Wnt, Notch, TGF-β, and EMT regulators create multistable systems that permit reversible phenotypic switching under stress. 12
Energy landscape modeling extends Waddington’s epigenetic landscape framework by describing cellular states as valleys separated by transition barriers of varying stability. In cancer, oncogenic signaling, epigenetic remodeling, and therapeutic stress reshape these landscapes, facilitating transitions into resistant or stem-like attractors. 73 Complementing these approaches, single-cell multiomics and lineage-tracing analyses enable reconstruction of cellular trajectories and transitional intermediates during therapy adaptation. 15
Bifurcation analysis further explains how small perturbations in signaling, metabolism, or chromatin accessibility can destabilize existing attractors and induce large-scale transcriptional reorganization into drug-tolerant states. 74 More recently, control theory-based frameworks have sought to identify regulatory “driver nodes” whose perturbation may redirect tumor cells toward differentiation or therapy-sensitive phenotypes. Together, these computational strategies strengthen the CSC attractor-state model by linking systems theory with predictive and potentially actionable therapeutic design.
Conclusion
The CSC paradigm has evolved from a rigid hierarchical doctrine into a dynamic regulatory framework that more accurately captures tumor complexity. Evidence from lineage tracing, single-cell, spatial multiomics, and systems biology indicates that stemness is now understood as a reversible and context-dependent cellular state embedded within a high-dimensional regulatory landscape. In this framework, CSCs can be interpreted as metastable attractor states shaped by interacting signaling networks, epigenetic regulation, metabolic programs, and microenvironmental inputs. Tumor heterogeneity and resistance arise through the interplay of clonal evolution, reversible state transitions, and niche-mediated stabilization. The CSC niche, comprising stromal, immune, vascular, metabolic, and mechanical components, further reinforces these dynamics through nonlinear regulatory coupling. Collectively, these findings support a shift from static, CSC-centric models toward ecosystem-based frameworks in which stemness is a reversible systems property. Future advances in single-cell, spatial, and perturbation-based technologies will enable the quantitative reconstruction of tumor attractor landscapes and the identification of regulatory control nodes capable of enforcing durable therapeutic collapse of adaptive cancer states.
Authors’ Contributions
The author contributed to the conception and design of the study, conducted the literature review, wrote the article, critically revised it, and approved the final version for submission.
Footnotes
Data Availability Statement
No new data were collected or analyzed.
Ethical Responsibilities
The author confirms that this study was conducted in accordance with established ethical standards. All information has been presented with accuracy, integrity, and transparency. The article is free from plagiarism, data fabrication, and all forms of unethical practice.
Author Disclosure Statement
The author declares that there are no conflicts of interest.
Funding Information
This research did not receive any specific funding from public, commercial, or nonprofit organizations.