
Abstract
Lung adenocarcinoma (LUAD) remains the most common subtype of lung cancer, characterized by high heterogeneity and poor survival outcomes. Although transcriptomic and metabolomic alterations have been individually studied, integrated multi-omics analyses are needed to uncover the convergent pathways that drive tumor progression. Differentially expressed genes (DEGs) were identified from the GSE229253 transcriptomic dataset comprising LUAD tumor and adjacent normal tissues, while significantly altered metabolites were obtained from the Lung Cancer Metabolome Database. The top 10 DEGs and metabolites were analyzed using the search tool for interacting chemicals (STITCH) to construct gene-metabolite networks, and Integrated Molecular Pathway Level Analysis (IMPaLA) was employed for integrated pathway enrichment to identify overlapping molecular processes. Transcriptomic profiling revealed 973 DEGs (410 upregulated and 563 downregulated), and metabolomic analysis identified significant alterations in metabolites linked to redox balance, amino acid derivatives, and nucleotide metabolism. Integration through STITCH generated a network of 16 nodes and 9 edges, highlighting gene-metabolite associations of probable biological relevance. Joint pathway enrichment analysis using IMPaLA consistently identified glycosylation-related pathways, particularly O-linked glycosylation of mucins, as major axes of convergence between transcriptomic and metabolomic alterations in LUAD (joint p = 0.00129–0.00434). Several genes (B3GNT6, FEZF1-AS1, and LCAL1) and metabolites (isoleucylleucine, leucylleucine, and isoleucylvaline) are probable novel candidates, warranting further investigation. These findings provide systems-level evidence that aberrant glycosylation is likely a central hallmark of LUAD, underscore the potential of glycosylation pathways as biomarkers and therapeutic targets, and demonstrate the utility of cross-omics approaches to unpack the molecular complexity of lung cancer.
Keywords
Introduction
Lung cancer remains the leading cause of cancer-related mortality worldwide, accounting for approximately 1.8 million deaths annually (Thandra et al., 2021). Among its histological subtypes, lung adenocarcinoma (LUAD) is the most prevalent, representing nearly 40% of cases (Lou et al., 2025). Despite advances in early detection and targeted therapies, the 5-year survival rate of LUAD patients remains below 20%, largely due to late-stage diagnosis, extensive heterogeneity, and limited availability of robust biomarkers. This underscores the urgent need to elucidate the molecular underpinnings of LUAD progression and to discover novel diagnostic and therapeutic targets.
Recent research highlights that cancer progression involves widespread reprogramming at both the transcriptomic and metabolomic levels (Dai et al., 2025; Gong et al., 2024). Altered gene expression profiles drive tumor initiation, proliferation, and metastasis, while metabolic reprogramming supports the energetic and biosynthetic demands of malignant cells (Nong et al., 2023). Transcriptomics provides a comprehensive view of deregulated signaling pathways, whereas metabolomics directly captures the biochemical consequences of these changes (Dasgupta et al., 2022; Choudhury et al., 2024). Both omics approaches, individually, have indeed advanced our understanding of LUAD, but integrating transcriptomic and metabolomic datasets is poised to bring about new vistas in understanding lung cancer biology. Multi-omics integration of transcriptomic and metabolomic findings can enable a systems-level perspective that links genetic dysregulation to metabolic phenotypes. Such integrative strategies are also valuable for identifying convergent molecular signatures that may otherwise remain undetected when each omics knowledge layer is analyzed in isolation from other omics platforms.
Integrated multi-omics approaches provide a comprehensive framework to unravel the molecular complexity of lung cancer by simultaneously capturing transcriptomic and metabolomic alterations (He et al., 2025). Transcriptomic profiling highlights dysregulated gene expression patterns that drive oncogenic signaling, immune modulation, and metabolic reprogramming, while metabolomic analysis reflects the downstream biochemical consequences of these transcriptional shifts. When integrated through pathway-level analyses, these complementary data layers enable a systems-level perspective—linking gene dysregulation with metabolite perturbations in shared biological processes such as nucleotide biosynthesis, redox regulation, and energy metabolism. Several studies have applied integrative omics approaches to cancer, combining transcriptomic, proteomic, or metabolomic datasets to uncover dysregulated pathways (Heo et al., 2021; Chakraborty et al., 2024). For example, network-based integration of gene expression and metabolite profiles has been used to identify biomarkers in several cancers, including breast and colorectal cancers, among others. (Hozhabri et al., 2022; Guo et al., 2017).
In this study, a cross-omics integration framework was applied to systematically investigate the interplay between dysregulated genes and metabolites in LUAD. The workflow presented in the present study differs from previous methods by specifically integrating top DEGs with significantly altered metabolites using search tool for interacting chemicals (STITCH)-based network analysis and integrated molecular pathway level analysis (IMPaLA) pathway enrichment. This enables systematic identification of convergent molecular pathways, such as aberrant glycosylation in LUAD, which has not been highlighted in prior integrative studies. Differentially expressed genes (DEGs) were identified from publicly available transcriptomic data, while significantly altered metabolites were obtained from curated publicly available lung cancer metabolome datasets. STITCH-based network analysis and IMPaLA pathway enrichment were employed to map interactions between gene expression and metabolic changes, enabling the identification of shared biological processes.
Materials and Methods
The present study employed publicly available omics data and did not require informed consent or institutional review board approval. The study was conducted under the overall research ethics oversight of the author’s institution.
Genes and metabolites were prioritized for integrative analysis based on their fold-change values, in combination with adjusted p values (false discovery rate (FDR), to focus on the most substantially dysregulated features. This strategy ensures that selected candidates represent both statistically significant and biologically meaningful alterations, improving the interpretability of downstream network and pathway analyses. By emphasizing features with larger dysregulation, the integrative analysis can more effectively capture key molecular pathways, such as glycosylation, that are perturbed in a disease state such as cancer.
Identification of differentially expressed genes
Transcriptomic data were retrieved from the GSE229253 dataset available in the NCBI Gene Expression Omnibus (GEO) database (Dolgalev et al., 2023). This dataset includes 33 lung tissue samples comprising 18 tumor tissues and 15 tumor-adjacent normal tissues from patients with LUAD. Sequencing was performed using the Illumina NovaSeq 6000 platform, generating high-throughput RNA-seq data. Differential gene expression analysis between tumor and normal tissues was carried out using standard statistical pipelines, and genes were considered significantly differentially expressed at adjusted p < 0.001 and |log2 fold change (FC)| ≥ 2. The GEO2R tool, with sample and control group definitions updated in November 2020, was used to identify the altered gene sets in the disease group.
Figure 1 illustrates the volcano, MA, box, Uniform Manifold Approximation and Projection (UMAP), and mean–variance plots, providing an overview of differential expression, normalization, clustering, and data quality assessment in LUAD compared to controls.
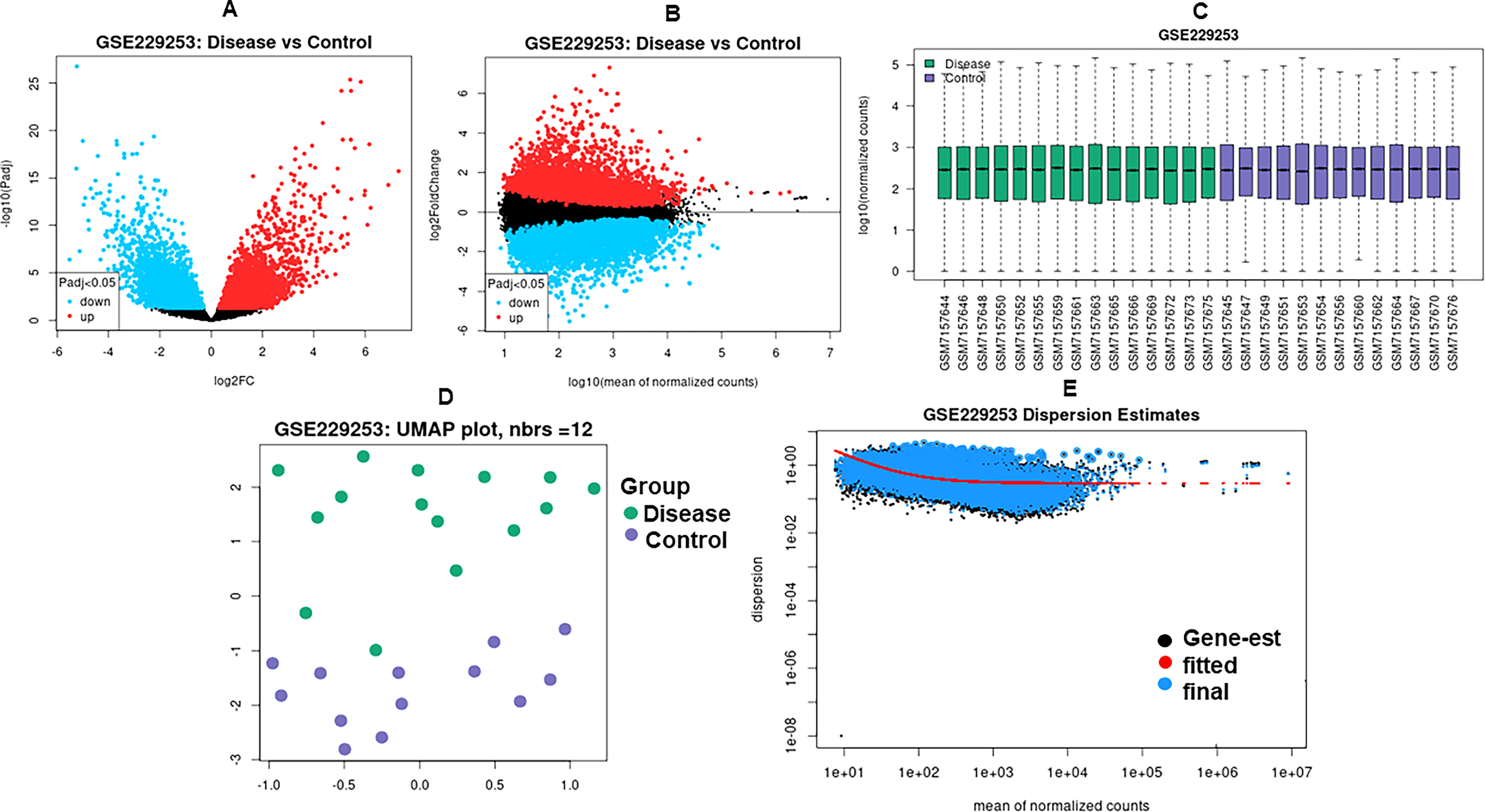
Exploratory and differential expression analyses of the LUAD dataset.
Metabolite screening
Metabolomic data were obtained from the Lung Cancer Metabolome Database (LCMD) (Wu et al., 2022), which includes paired tumor and adjacent normal lung tissue samples from patients with adenocarcinoma (n = 33) and squamous cell carcinoma (n = 35) at stages I–III. Samples were analyzed using liquid chromatography (LC) and gas chromatography (GC) coupled with electrospray ionization and electron ionization, followed by mass analysis using linear ion-trap (LC) and single-quadrupole (GC) systems. Metabolite identification was confirmed using Liquid Chromatography with tandem mass spectrometry (LC—MS/MS). Differential metabolites were determined using paired two-sample t-tests and Partial Least Squares Discriminant Analysis (PLS-DA) with multiple testing correction (FDR < 0.05) (Moreno et al., 2018). Annotation was supported by KEGG and HMDB databases.
Construction of metabolite-gene networks
To explore functional interactions between significantly altered genes and metabolites, the present study selected the top 10 DEGs from the transcriptomic dataset and the top 10 significantly dysregulated metabolites from the metabolomic dataset. These were integrated using the STITCH database (Search Tool for Interactions of Chemicals and Proteins, v5.0), which curates experimentally validated and computationally predicted associations between small molecules and proteins (Kuhn et al., 2008).
The input list of gene symbols and metabolite names was queried against the Homo sapiens background. Associations were retrieved based on text mining, experimental evidence, curated databases, and co-expression information, with a confidence score threshold of 0.4 (medium confidence). The resulting network consisted of nodes (representing genes or metabolites) and edges (representing interactions between entities).
Pathway enrichment and integration
To identify common biological processes perturbed at both transcriptomic and metabolomic levels, the present study performed integrative pathway enrichment analysis using IMPaLA: integrated molecular pathway level analysis (http://impala.molgen.mpg.de/) (Kamburov et al., 2011). IMPaLA allows for the simultaneous evaluation of gene and metabolite datasets within a unified pathway context.
The list of significantly DEGs and the top significantly altered metabolites was uploaded as input. Overrepresentation analysis was then conducted separately for genes and metabolites against multiple curated pathway databases, including Reactome Pathway Knowledgebase (Reactome) and the Edinburgh Human Metabolic Network (EHMN).
For each pathway, IMPaLA computes:
p-Value (genes): statistical significance of enrichment based on the gene set. p-Value (metabolites): enrichment significance for the metabolite set. Joint p-value: integrated probability score reflecting the combined evidence from both gene and metabolite enrichment. Q-values: FDR-adjusted values to correct for multiple hypothesis testing.
Pathways were considered significantly enriched if the joint p-value < 0.05 following FDR correction. This approach enabled us to pinpoint overlapping gene-metabolite pathways, thereby uncovering molecular mechanisms jointly supported by transcriptomic and metabolomic evidence in LUAD. In addition, to evaluate the robustness of the findings, exploratory analyses were initially conducted with the top 10 DEGs and metabolites and were repeated using the top 50 DEGs and metabolites.
Results
Differential gene expression in LUAD
Transcriptomic profiling of the GSE229253 dataset revealed 973 DEGs, including 410 upregulated and 563 downregulated genes in LUAD tumor tissues compared to adjacent controls. The most significantly dysregulated genes included:
Upregulated: NELL1 (log2FC = 7.30), LCAL1 (log2FC = 6.90), B3GNT6 (log2FC = 6.21), FEZF1-AS1 (log2FC = 6.16), SYT12 (log2FC = 6.09). Downregulated: MT1A (log2FC = –5.52), CD5L (log2FC = –5.24), HAS1 (log2FC = –5.13), FOLR3 (log2FC = –5.00), CXCR2 (log2FC = –4.97).
These results indicate substantial transcriptomic reprogramming, particularly in glycosylation-related and immune-regulatory genes. Table 1 summarizes the top ten DEGs ranked by log2 FC in LUAD compared to controls.
Summary of the Top 10 Differentially Expressed Genes Ranked by log2 Fold Change in LUAD Compared to Controls

Altered metabolomic profile in LUAD
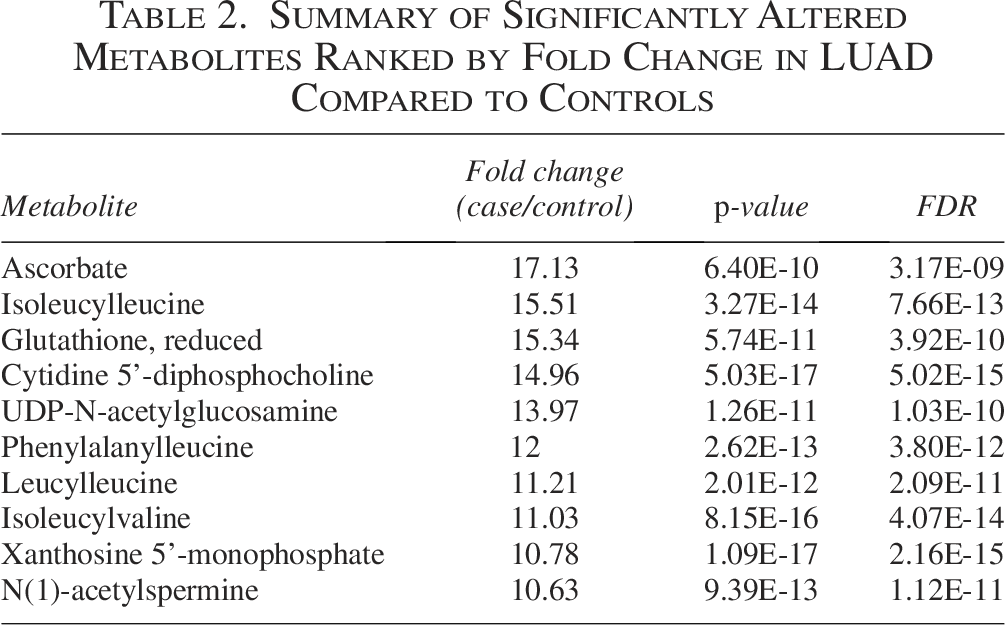
Metabolomic analysis identified a panel of significantly altered metabolites between tumor and adjacent normal lung tissues. The top 10 metabolites ranked by FC included antioxidants, amino acid derivatives, nucleotide intermediates, and polyamine metabolites. Among these, ascorbate (FC = 17.13, FDR = 3.17 × 10−9) and reduced glutathione (FC = 15.34, FDR = 3.92 × 10−10) were markedly elevated, suggesting enhanced redox modulation in LUAD. Nucleotide metabolism intermediates such as cytidine 5′-diphosphocholine (FC = 14.96, FDR = 5.02 × 10−15) and xanthosine 5′-monophosphate (FC = 10.78, FDR = 2.16 × 10−15) were also significantly enriched. Table 2 summarizes the metabolites ranked by FC in LUAD compared to controls.
Summary of Significantly Altered Metabolites Ranked by Fold Change in LUAD Compared to Controls
Metabolite-gene network analysis
The STITCH-based integrative network combining the top 10 DEGs and top 10 significantly altered metabolites in LUAD yielded a network consisting of 16 nodes (representing genes and metabolites) and 9 edges (representing predicted or validated associations). The network exhibited an average node degree of 1.12, indicating that most entities were connected to at least one partner, although highly connected hubs were limited. The clustering coefficient was 1, suggesting that the connected nodes formed tightly closed triplets, reflecting localized modular interactions.
The expected number of edges was seven, compared to the observed nine, which demonstrates a modest enrichment of cross-omics connectivity. The protein–protein interaction (PPI) enrichment p-value of 0.328 indicated that the overall number of associations was not significantly higher than expected by chance, yet the specific connections identified—particularly those linking glycosylation-related genes with metabolites involved in amino sugar and nucleotide metabolism—may represent biologically meaningful regulatory interactions.
Collectively, the network analysis highlights a subset of metabolite-gene interactions that converge on critical cellular processes, suggesting that even a modest degree of connectivity between dysregulated transcripts and metabolites can reveal functionally relevant cross-omics signatures in LUAD (Fig. 2).

Network statistics of the LUAD-associated interaction network. The constructed protein–protein interaction (PPI) network comprised 16 nodes and 9 edges, with an average node degree of 1.12 and a clustering coefficient of 1. The expected number of edges was 7, and the observed connectivity did not show significant enrichment (PPI enrichment p-value = 0.328). LUAD, lung adenocarcinoma.
Integrated pathway enrichment
Joint pathway analysis using IMPaLA (http://impala.molgen.mpg.de/) revealed significant convergence between the transcriptomic and metabolomic datasets in LUAD. Among the enriched pathways, the strongest overlaps were consistently observed in glycosylation-related processes, highlighting their central role in tumor biology.
O-linked glycosylation of mucins: driven by the upregulated gene B3GNT6 and the elevated metabolite Uridine diphosphate (UDP)-N-acetylglucosamine, indicating aberrant mucin-type glycan extension in LUAD. O-glycan biosynthesis: enriched through the same gene-metabolite pair, underscoring altered initiation and elongation of O-glycan chains. O-linked glycosylation: overlapping via B3GNT6 and UDP-N-acetylglucosamine, further confirming the disruption of glycosylation machinery.
The joint enrichment p values (0.00129–0.00434) provide robust statistical support for the integration of transcriptomic and metabolomic signals within these pathways. These findings suggest that aberrant O-glycosylation represents a major axis of transcriptome–metabolome convergence in LUAD, potentially contributing to altered cell–cell communication, immune evasion, and extracellular matrix remodeling during tumor progression. Table 3 highlights the top three enriched pathways identified through IMPaLA analysis in LUAD.
Enriched Glycosylation-Related Pathways Identified by IMPaLA Analysis in LUAD

LUAD, lung adenocarcinoma
Expansion of the feature set to the top 50 DEGs and metabolites confirmed the robustness of the pathway-level findings. The names of these features are provided in Supplementary Table S1. IMPaLA enrichment analysis again highlighted O-linked glycosylation and related pathways as among the most significantly enriched, with joint p values of 1.85 × 10−7 (q = 0.0001) for O-linked glycosylation (Supplementary Table S2). This consistency across feature-set sizes indicates that the glycosylation signal is not dependent on the initial top-10 cut-off.
Discussion
The integrative analysis of transcriptomic and metabolomic datasets in LUAD unpacks and evaluates the importance of glycosylation-related pathways, particularly O-linked glycosylation of mucins, as central nodes of convergence. In the present study, the identification of B3GNT6 as a key upregulated gene, along with elevated levels of UDP-N-acetylglucosamine, suggests that aberrant glycosylation may play a pivotal role in the molecular reprogramming of LUAD. O-glycosylation of mucins is known to influence cell–cell and cell–matrix interactions, facilitate immune evasion, and promote metastatic potential (Bhatia et al., 2019). The current findings provide multi-omics evidence supporting these processes as critical drivers of lung tumor progression.
Metabolomic profiling in LUAD demonstrated pronounced upregulation of metabolites involved in redox homeostasis (ascorbate, reduced glutathione), nucleotide metabolism (cytidine 5′-diphosphocholine, xanthosine 5′-monophosphate), and polyamine biosynthesis [N(1)-acetylspermine]. The elevation of antioxidants reflects the adaptive response of tumor cells to increased reactive oxygen species (ROS), a hallmark of the oxidative stress environment in LUAD (Attique et al., 2025). The accumulation of ROS contributes to multiple disorders beyond the lungs, including reproductive dysfunction (Singh et al., 2022) and chronic kidney disease (Irazabal and Torres, 2020). Concurrently, enhanced nucleotide intermediates support the heightened demand for DNA synthesis and transcription required for uncontrolled cellular proliferation (Mullen and Singh, 2023). The accumulation of polyamine derivatives further underscores the metabolic reprogramming that promotes tumor growth and survival, positioning these pathways as central metabolic signatures of LUAD progression.
The concurrent dysregulation of transcriptomic profiles, particularly genes implicated in glycosylation (B3GNT6) and immune modulation (CXCR2 and CD5L), points to a coordinated reprogramming of molecular networks in LUAD. Aberrant glycosylation can remodel the tumor microenvironment by influencing cell–cell adhesion, signaling, and immune evasion, while altered immune-related gene expression promotes chronic inflammation and suppression of anti-tumor immunity (Peixoto et al., 2019). Together, these transcriptomic alterations reinforce the metabolic rewiring observed at the metabolite level, highlighting a synergistic framework that facilitates tumor survival, proliferation, and disease progression.
The network analysis conducted through STITCH demonstrated modest but biologically relevant gene-metabolite associations, reinforcing the concept that key interactions rather than dense connectivity may drive tumor-specific phenotypes. Although the overall PPI enrichment was not statistically significant, the presence of directed links between glycosylation-related genes and nucleotide-sugar metabolites underscores the biological plausibility of the observed pathways. This highlights the advantage of cross-omics approaches, where even limited overlap between molecular layers can reveal mechanistic insights that would not be apparent in single-omics analyses.
The enrichment of glycosylation pathways is consistent with prior studies showing that aberrant glycan structures are frequently observed in LUAD and contribute to altered signaling, metastasis, and therapeutic resistance (Xu et al., 2024; Zhang et al., 2025). IMPALA-based integration highlighted multiple glycosylation-related pathways (Table 3), all converging on the interaction between B3GNT6 and UDP-N-acetylglucosamine. Although these pathways are not independent, their repeated enrichment across Reactome and EHMN repositories emphasizes glycosylation as a consistent and probable hallmark of LUAD.
Importantly, the integration of transcriptomic and metabolomic evidence provides stronger support for the role of O-linked glycosylation as a hallmark of LUAD biology. Given that glycosyltransferases and glycan intermediates are pharmacologically targetable, the findings may inform the development of glycosylation-focused biomarkers and therapeutic interventions. To reinforce the biological plausibility of the findings, pathway-level sensitivity analyses were conducted using expanded feature sets (top 50 DEGs and metabolites). These analyses consistently identified glycosylation-related pathways, including O-linked glycosylation of mucins, among the most significantly enriched terms (joint p = 1.85 × 10−7, q = 0.0001; Supplementary Table S1). The reproducibility of glycosylation enrichment across feature-set sizes supports the robustness of the signal despite limited overall network significance.
Beyond mechanistic involvement in LUAD progression, glycosylation alterations also carry translational significance. Several glycosyltransferases represent druggable nodes, including GALNT family enzymes that initiate O-glycosylation, FUT8, which mediates core fucosylation and has been linked to epidermal growth factor receptor (EGFR) signaling, and B3GNT family members such as B3GNT6 identified in the present analysis. Inhibitors and RNAi-based approaches directed against these enzymes are under preclinical evaluation. Mucin-directed therapeutics also represent an emerging strategy: antibodies that recognize tumor-associated glycoforms of MUC1 (e.g., SAR566658, gatipotuzumab) and mucin-based vaccines are being tested in clinical settings (Nicolazzi et al., 2020; Grewal and Kurzrock, 2025). Furthermore, glycosylation-derived biomarkers such as serum MUC1 glycoforms and elevated FUT8 expression have shown potential prognostic and predictive value in LUAD. Integration of transcriptomic and metabolomic evidence with these translational developments emphasizes glycosylation pathways as not only hallmarks of disease biology but also promising targets for precision diagnostics and therapeutics.
Several limitations of the present study are noteworthy. First, the transcriptomic dataset analyzed was limited to a moderate sample size, and external validation in larger independent cohorts would strengthen confidence in the results. Second, the metabolomic data were obtained from curated repositories such as the LCMD, which may introduce potential biases due to differences in annotation depth, metabolite coverage, and reliance on prior knowledge for metabolite identification. Second, the samples included in the analysis spanned stages I–III of LUAD, introducing heterogeneity that could confound pathway-level interpretations. While this heterogeneity reflects the clinical diversity of the disease, it also underscores the need for stage-stratified analyses in future studies. Addressing these limitations through validation in larger, independent, and stage-matched cohorts will be critical to strengthening the translational applicability of the integrative findings. Third, the integration was restricted to genes and metabolites with the strongest FCs; thus, subtle but biologically relevant signals may have been overlooked. Fourth, a single well-annotated transcriptomic dataset (GSE229253) and a corresponding metabolomic dataset from LCMD were selected to ensure methodological consistency and minimize variability arising from sequencing platforms, sample processing, and clinical annotation. Although incorporating multiple datasets could increase statistical power, the current approach enabled a controlled, initial evaluation in regard to the integration of transcriptomic and metabolomic layers of data in LUAD. Future studies should aim to include multiple independent cohorts, apply batch correction and normalization, and validate the identified gene-metabolite interactions and pathway signatures to strengthen the robustness, generalizability, and translational applicability of the findings. Despite these limitations, the convergence of transcriptomic and metabolomic evidence on glycosylation pathways provides a compelling framework for future mechanistic and translational studies. Furthermore, future research should aim to extend these findings by validating the identified transcriptomic–metabolomic associations in larger and independent LUAD cohorts, thereby strengthening their translational significance. Incorporating additional omics layers, such as proteomics and epigenomics, could further enhance the resolution of molecular networks and capture regulatory events that bridge gene expression and metabolic output. Moreover, next-generation phenomics, leveraging emerging single-cell and spatial multi-omics technologies, extends beyond cross-omics integration to dissect intratumoral heterogeneity, map cell type-specific contributions, and uncover microenvironmental interactions driving disease progression (Dasgupta, 2024). Integration of such high-dimensional datasets within precision oncology frameworks may ultimately facilitate the development of more robust biomarkers and tailored therapeutic strategies for LUAD.
Conclusions and Outlook
In all, the integrative transcriptomic–metabolomic analysis underscores aberrant glycosylation as a probable hallmark of LUAD. Ten key genes were identified herein, including NELL1, LCAL1, B3GNT6, FEZF1-AS1, SYT12, MT1A, CD5L, HAS1, FOLR3, and CXCR2, alongside 10 key metabolites, namely ascorbate, isoleucylleucine, reduced glutathione, cytidine 5′-diphosphocholine, UDP-N-acetylglucosamine, phenylalanylleucine, leucylleucine, isoleucylvaline, xanthosine 5′-monophosphate, and N(1)-acetylspermine. The results point toward several genes (such as B3GNT6, FEZF1-AS1, and LCAL1) and metabolites (including isoleucylleucine, leucylleucine, and isoleucylvaline) that are probable novel candidates requiring further investigation. Pathway-level integration highlighted glycosylation processes, particularly O-linked glycosylation of mucins, as probable convergence points between gene expression and metabolic alterations. While these findings require validation in independent and stage-stratified cohorts, they provide a framework for future studies aimed at refining biomarker discovery and identifying therapeutic opportunities in LUAD.
Looking further, this integrative cross-omics analysis highlights the convergence of transcriptomic and metabolomic dysregulation in LUAD, underscoring glycosylation and redox balance as potentially central molecular axes of disease progression. The interplay between altered metabolites, including antioxidants and nucleotide precursors, and dysregulated genes involved in glycosylation and immune modulation suggests a coordinated reprogramming that sustains tumor proliferation and survival.
By linking metabolic adaptation with gene-driven remodeling of the tumor microenvironment, this study provides new mechanistic insights into LUAD biology. Importantly, the findings emphasize glycosylation pathways as potential biomarkers and therapeutic targets, while also demonstrating the utility of integrated omics approaches for advancing precision oncology.
Footnotes
Acknowledgments
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for the present study.
Author’s Contributions
S.D.: Conceptualization, formal analysis, investigation, visualization, and writing—review/editing.
Supplemental Material
Supplemental Material
Abbreviations
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.