
Abstract
Lentinan (LNT) is a polysaccharide with antioxidant and anti-inflammatory properties; however, its link to intervertebral disc degeneration (IVDD) remains unclear. Here, we integrated network pharmacology, machine learning, molecular docking, and molecular dynamics (MD) simulations to investigate the LNT–IVDD relationship. Using dataset GSE176205, 16,361 potential IVDD-related genes were identified, and 97 LNT-associated targets were collected from four databases, yielding 46 intersecting targets. GO and KEGG analyses revealed enrichment in pathways including calcium, PI3K-Akt, Notch, and Rap1 signaling. Protein–protein interaction network analysis combined with three ML methods prioritized two core targets: signal transducer and activator of transcription 3 (STAT3) and fibroblast growth factor 2 (FGF2). The diagnostic relevance of these genes was subsequently validated using receiver operating characteristic curves across three independent IVDD datasets. Molecular docking showed strong binding (energy < −5 kcal/mol) between LNT and these targets, further supported by stable complex formation in MD simulations. Immune-infiltration analysis delineated the IVDD microenvironment, and virtual knockout based on single-cell transcriptomics systematically assessed how perturbing key genes remodels cell states and signaling networks. Together, this work provides novel insights into the molecular mechanisms underlying LNT’s therapeutic effects in IVDD and establishes a foundation for its translational study.
Keywords
Introduction
Intervertebral disc degeneration (IVDD) is a common degenerative musculoskeletal disease and the leading cause of chronic low back pain, which severely compromises the quality of life of those affected. In addition to the personal suffering, IVDD places a considerable economic burden on society (GBD 2019 Diseases and Injuries Collaborators, 2020; Juch et al., 2017). The degenerative process is mediated by a cascade of pathological events, principally involving the loss of nucleus pulposus (NP) cells, a loss of structural integrity in the annulus fibrosus (AF), and the breakdown of the extracellular matrix (ECM) (Ding et al., 2013; Molladavoodi et al., 2020). Evidence indicates that nearly 70% of adults will experience low back pain at some point in their lives (Liu et al., 2023). Currently, the typical treatment options for IVDD mainly consist of nonsurgical methods, such as pain relief medications and physical therapy, as well as surgical procedures when required (Velnar and Gradisnik, 2023). However, conservative treatments generally provide only temporary symptomatic relief and are ineffective in restoring the disc microenvironment or halting disease progression (Santos et al., 2022). Although surgical intervention can successfully alleviate mechanical compression of nerve roots caused by a herniated NP, it typically does not reverse the underlying degenerative process or restore the native disc’s structure and function (Daly et al., 2016). Notably, oxidative stress and metabolic imbalance play central roles in driving NP cell dysfunction and ECM breakdown (Chen et al., 2024).
Lentinan (LNT) is a polysaccharide extracted from the shiitake mushroom, whose primary structure is a β-1,3-glucan. It exhibits a broad spectrum of therapeutic properties, including immunomodulatory, anti-inflammatory, antioxidant, and antitumor activities (Chihara et al., 1969; Meng et al., 2022; Song et al., 2024; Stellavato et al., 2016; Wang et al., 2020; Wu et al., 2021). It functions as a biological response modifier, enhancing immune cell activity and mitigating oxidative and inflammatory damage (Kaleta et al., 2019; Liu et al., 2019; Ooi and Liu, 2000; Ren et al., 2018; Shirasaka et al., 1996). However, its role in IVDD remains underexplored, particularly concerning its multitarget mechanisms and therapeutic applicability.
Network pharmacology is instrumental in elucidating the multitarget mechanisms of bioactive compounds (Hopkins, 2007). This methodology focuses on elucidating complex interaction networks involving drugs, biological pathways, molecular targets, and disease processes, emphasizing the inherent polypharmacology of many therapeutic interventions (Hopkins, 2008). Molecular docking is a core technique in structural biology for simulating ligand–target interactions and evaluating binding affinity. The strength of this interaction is quantified by the binding energy, with lower values indicating enhanced complex stability and a higher probability of successful binding (Li et al., 2022). To analyze the stability and time-dependent evolution of biological complexes, molecular dynamics (MD) simulations are frequently employed. By modeling protein–ligand interactions within a near-physiological environment, MD provides critical information on complex stability and conformational changes (Wu et al., 2022). In this study, MD simulation was further employed to validate the interaction modes and structural stability of the LNT–target complexes predicted by molecular docking, offering comprehensive dynamic insights for potential pharmacological applications.
This study adopted an integrated, multimethod strategy to construct a research framework aimed at elucidating the underlying pharmacological mechanisms of LNT against IVDD. The initial step of our analytical pipeline was the acquisition of differentially expressed genes (DEGs) associated with IVDD from the GSE176205 dataset. Concurrently, potential therapeutic targets of LNT were retrieved from various databases, including Herb (Fang et al., 2021), Swiss Target Prediction (Daina et al., 2019), GeneCards (Stelzer et al., 2016), and SEA (Keiser et al., 2007). An integrated “drug–target–disease” network was established to delineate the interrelationships based on the acquired data. Through network pharmacology techniques, we systematically pinpointed the core signaling pathways and biological processes (BPs) through which LNT exerts its effects on IVDD. Three specific algorithms—least absolute shrinkage and selection operator (LASSO), random forest (RF), and extreme gradient boosting (XGBoost)—were employed for feature selection to optimize the identification of key targets and improve the model’s predictive reliability. Building on this foundation, we utilized molecular docking and MD simulations to comprehensively validate the binding strength and conformational stability of LNT with the core targets, evaluating both static binding conformations and dynamic interactions. Furthermore, immune infiltration analysis was employed to delineate the immune microenvironment of IVDD. Leveraging single-cell transcriptomic data, we conducted virtual gene knockout experiments to systematically assess the impact of perturbing key genes on cellular state transitions and the remodeling of signaling pathways. From multiomics, molecular docking, dynamic simulation, and cellular functional perspectives, this study systematically elucidates the molecular mechanisms through which LNT mitigates IVDD, thereby providing a theoretical foundation for its clinical translation from symptom alleviation to disease modification.
Materials and Methods
IVDD data collection and differential expression analysis
Within the gene expression omnibus (GEO) repository, a systematic search using the term “Intervertebral Disc Degeneration” retrieved the dataset GSE176205, which is based on the GPL20301 platform (Supplementary Table S1). From this dataset, we selected three samples from healthy controls and five samples from patients with IVDD for further analysis. To minimize nonbiological technical variations, such as batch effects and platform-related biases, the raw microarray dataset was normalized utilizing the normalizeBetweenArrays() function implemented in the limma R package (version 4.5.0). This step ensured that the expression values were standardized across samples, allowing for accurate comparisons. Differential expression analysis was carried out by comparing the gene expression profiles between healthy control and IVDD samples. We compared gene expression between the healthy control and IVDD groups, with the criteria for identifying significant DEGs set at an absolute |log2FC| > 1 and an adjusted p value < 0.05. Principal component analysis (PCA) was subsequently conducted on the Bioinformatics online platform to visually assess global gene expression patterns and examine sample clustering (Tang et al., 2023). To further depict the distribution and expression characteristics of the DEGs, volcano plots and heatmaps were constructed.
Comprehensive identification of therapeutic targets for LNT
To identify potential targets of LNT, its SMILES identifier was first obtained from the Herb database. The Herb, Swiss Target Prediction, GeneCards, and SEA databases were searched using this identifier for the term “Lentinan,” restricting the focus to human (Homo sapiens) targets (Supplementary Table S1). Following structural verification and consistency evaluation of the retrieved results, a consolidated dataset of high-confidence potential targets was constructed. Common targets between LNT and IVDD were identified using Venn diagram analysis. The overlapping targets were defined as potential therapeutic targets through which LNT may exert effects on IVDD.
Enrichment characterization of target protein functions and pathways
The core set of genes was interrogated through functional annotation and enrichment analysis, using the Metascape platform to decipher underlying molecular mechanisms and pathway regulations systematically (Supplementary Table S1). Initially, the list of intersecting targets was input into Metascape, followed by the execution of a standard enrichment workflow under the “Express Analysis” mode. Our enrichment analysis encompassed annotations from the major gene ontology (GO) classifications (BP, cellular component [CC], and molecular function [MF]) as well as pathways from the kyoto encyclopedia of genes and genomes (KEGG) database (Gene Ontology Consortium, 2015; Kanehisa et al., 2017). Subsequently, the enrichment results were sorted in ascending order based on adjusted p values to prioritize the most significant biological functions. From these, the top 10 GO terms (BP, CC, and MF) and the top 20 KEGG pathways were selected for in-depth interpretation. To improve the interpretability and presentation of the results, key GO terms and KEGG pathways were graphically represented through the Sangerbox and Bioinformatics online tools, respectively (Chen et al., 2024; Tang et al., 2023).
Building protein–protein interaction (PPI) network to identify hub targets
We employed the STRING database to generate a PPI network for the intersecting targets, setting the organism to Homo sapiens and the interaction score threshold to 0.400 (Supplementary Table S1). Following this, the topological parameters of the resulting PPI network—including but not limited to degree centrality, betweenness centrality, and clustering coefficient—were systematically calculated and analyzed within Cytoscape utilizing the NetworkAnalyzer module (Shannon et al., 2003). To robustly identify pivotal regulatory targets within the network, an integrated multialgorithmic strategy was implemented through the combined application of CytoHubba, CytoNCA, and MCODE plugins. Core regulatory targets were rigorously defined as the consensus genes emerging from the intersection of the outputs generated by all three algorithms, thereby enhancing the reliability and biological relevance of the identified key nodes.
Integrated machine learning (ML) for screening core targets
This study analyzed 46 overlapping candidate genes using three well-established ML algorithms: LASSO, RF, and XGBoost. To ensure full reproducibility of all analyses, a fixed random seed of 123 was used throughout the study.
For binary classification, LASSO regression analysis was implemented in R (version 4.5.0) using the glmnet package, with the “family” parameter set to “binomial.” The optimal regularization parameter (λ) was determined by selecting the minimum value (λ.min) from cross-validation results. The trajectories of coefficient estimates for all features across a range of log(λ) values were visualized. Plotting the partial likelihood deviance (binomial deviance) against log(λ) provided a means to assess the model’s fit and regularization behavior.
To identify key predictors, a RF algorithm was implemented using the randomForest package (version 4.5.0) in R (Ellis et al., 2014). The model was built using 500 trees on the discovery cohort, with the optimal tree number chosen to minimize cross-validation error. Genes were ranked according to their importance scores, and the five most predictive genes were highlighted visually.
We employed the xgboost package in R to carry out the XGBoost analysis. The analysis involved calculating the receiver operating characteristic (ROC) curve and the area under the curve (AUC) to evaluate model performance. Feature importance scores were derived using the xgb.importance() function to evaluate the contribution of each gene to the model’s predictions. The most significant features were visualized using the plot() function. After applying these three algorithms, the union of all identified targets was compiled, and this gene set was designated as potential core targets for LNT and IVDD.
Finally, to define the core regulatory targets, genes identified using the three Cytoscape plugins were intersected with those derived from the ML analyses.
The datasets GSE15227, GSE70362, and GSE124272 were selected as validation sets for evaluation. To assess the discriminative ability of the identified core genes across different IVDD datasets, this study utilized the R package pROC to generate ROC curves. This approach systematically evaluated the diagnostic potential of the identified core targets and the robustness of the model. The AUC ranges from 0.5 to 1.0, with higher AUC values indicating better discriminative performance.
Molecular docking and MD simulation study on core targets of LNT
We performed molecular docking of LNT against the core targets to forecast their binding behavior and interaction patterns, which provided insights into the underlying mechanistic basis. Before docking, the crystal structure of the core target protein (sourced from the RCSB PDB) was prepared using PyMOL by deleting all water molecules and native ligands (Supplementary Table S1). Following its acquisition from PubChem, the 2D structure of LNT was subsequently evaluated for its minimum binding energy with Chem 3D (Supplementary Table S1). The protein structure was prepared for docking using AutoDockTools 1.5.7. Docking simulations between LNT (ligand) and the target protein (receptor) were carried out using AutoDock Vina in AutoDockTools to assess the binding stability. The results were visualized with PyMOL software.
The initial system for MD simulations, prepared in GROMACS 2023.2, was derived from the optimal docking pose as determined by a comprehensive evaluation of its binding energy, conformational clustering, and geometric complementarity. Using UCSF Chimera, the complex was subsequently dissociated into the protein receptor and the small molecule ligand. The ligand then underwent hydrogenation to optimize its protonation state and charge distribution, after which the adjusted ligand structure was submitted to the SwissParam server for parameterization. Concurrently, hydrogen atoms were added to the receptor protein, side-chain conformations were optimized, and nonessential water molecules were removed, all within the Chimera environment. Simulations were carried out in the NPT ensemble (300 K, 1 bar). For the energy calculations, the CHARMM27 force field was employed in conjunction with the explicit TIP3P water model. To achieve electrostatic neutrality, physiological concentrations of Na+ and Cl- ions were introduced into the system. The system underwent energy minimization via the steepest descent algorithm, followed by equilibration under both number of particles, volume, temperature ensemble (NVT) and number of particles, pressure, temperature ensemble (NPT) conditions. Production MD simulations were then executed employing a time step of 2 fs for a total duration of 50 ns. Trajectory analysis was performed using GROMACS built-in tools to assess key structural and dynamic properties, including root mean square deviation (RMSD) for overall stability, root mean square fluctuation (RMSF) to evaluate residue flexibility, and radius of gyration (Rg) to monitor global compactness. The binding-free energies were estimated using the MM-PBSA/GBSA method. In addition, free energy landscape analysis was conducted to identify metastable states and transition pathways. All structural and energetic data were visualized with DuIvyTools to aid in interpretation and presentation.
Immune cell infiltration analysis
Cibersort is a deconvolution algorithm based on linear support vector regression, which enables the inference of immune cell composition from bulk RNA sequencing or microarray expression data. It estimates the relative abundances of diverse human immune cell subtypes, thereby facilitating the characterization of immune cell infiltration profiles in transcriptomic datasets (Newman et al., 2015). In this study, the CIBERSORT algorithm was utilized to systematically characterize and compare the immune cell infiltration profiles between IVDD samples and healthy controls. The analysis was conducted with 1000 permutations, and quantile normalization was applied for proper adaptation to microarray data.
In silico FGF2 and STAT3 knockout identifies downstream regulatory networks
The GSE230808 dataset, comprising 18 IVDD samples, was retrieved from the GEO database. Single-cell RNA sequencing data were extracted and systematically analyzed using the Seurat pipeline. Raw expression matrices were log-normalized, and batch effects stemming from sample sources were corrected using the Harmony algorithm. Subsequently, uniform manifold approximation and projection (UMAP) was applied for nonlinear dimensionality reduction and visualization of cellular distribution. Following clustering, the resultant cell subpopulations were annotated via the online platform Annotation of Cell Types to characterize their respective identities (Supplementary Table S1).
To further investigate the potential regulatory functions of the core hub genes fibroblast growth factor 2 (FGF2) and signal transducer and activator of transcription 3 (STAT3) at the single-cell level, virtual knockout analysis was conducted utilizing the “scTenifoldKnk” R package (Osorio et al., 2022). A gene regulatory network was constructed to simulate the effects of target gene knockout, leading to the identification of a set of perturbed genes. Subsequent functional enrichment analysis of these genes was performed to systematically elucidate their involvement in BPs and signaling pathways.
Results
Systematic identification of LNT and IVDD targets
The overall analytical workflow is summarized in Figure 1a, including DEG identification, functional enrichment, PPI network construction, ML prioritization, consensus target selection, and external dataset validation. The dataset GSE176205, associated with IVDD, was obtained from the GEO database for in-depth bioinformatic analysis to uncover potential therapeutic targets. The data, comprising three healthy controls and five IVDD cases, were standardized (Fig. 1b). We then applied PCA to the multivariate data from both the healthy control and IVDD groups, aiming to reduce dimensionality and visually highlight differences between the two groups. The PCA results revealed a distinct separation between the groups (Fig. 1c).
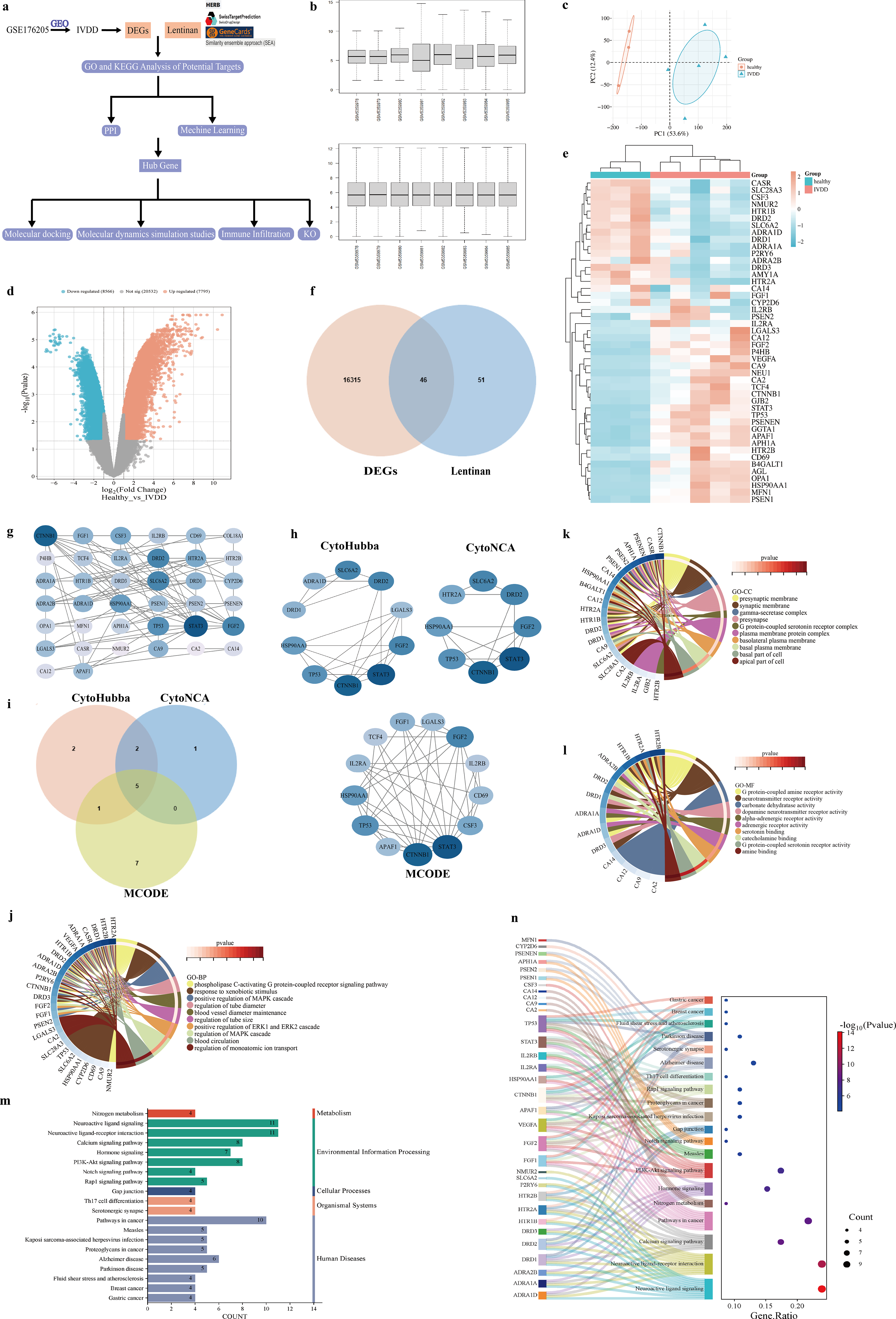
Differential analysis conducted with the limma package identified 16,361 genes that met the threshold of |log2FC| > 1 and an adjusted p value < 0.05. Compared with the control group, 7795 genes were significantly upregulated, while 8566 genes were downregulated in IVDD samples (Fig. 1d). A standardized retrieval strategy using four databases (Herb, Swiss Target Prediction, GeneCards, and SEA) identified 97 LNT-related genes. Through Venn analysis, 46 shared targets between LNT and IVDD were identified (Fig. 1f). The Venn diagram clearly and intuitively displays the sets of potential LNT targets and IVDD-related targets, as well as their intersection. Combined with the visualization of a volcano plot, the significant changes in expression levels of these overlapping targets are further highlighted (Fig. 1e).
Construction of a PPI network and identification of key hub genes for potential targets
We utilized the STRING database to build a PPI network consisting of 45 nodes and 100 interaction edges. Following its construction, the network’s topological properties were analyzed, and an optimized visualization was generated with Cytoscape (Fig. 1g). The size and color of the nodes correspond to their degree value. Larger size and darker color denote a higher degree, indicating greater functional importance within the network. Through the combined application of three Cytoscape plugins—CytoHubba, CytoNCA, and MCODE—screening was conducted from different dimensions to identify key genes (Fig. 1h). The intersection of results from these three methods yielded five core genes: CTNNB1, TP53, HSP90AA1, STAT3, and FGF2. Each of these genes exhibits high topological importance within the network, serves as a modular hub, and possesses critical biological functions (Fig. 1i; Supplementary Table S2).
Functional and pathway analysis of potential targets via GO and KEGG
Using the Metascape database, GO functional enrichment was carried out for the 46 potential targets, setting Homo sapiens as the biological model. The analysis identified 627 statistically significant GO terms, distributed as 521 in BP, 58 in CC, and 48 in MF. Based on p value ranking, the 10 most significant entries from each of the three GO categories were chosen for graphical representation (Fig. 1j–l). Furthermore, KEGG pathway enrichment analysis was performed on the 46 potential targets using the Metascape database to identify their involved specific signaling pathways. Among the total of 45 enriched signaling pathways, we generated a histogram of significant categories and a statistical bubble chart to visually display the top 20 significantly enriched KEGG pathways, ranked by p value (Fig. 2m,n).

Identification of core target genes for LNT and IVDD through three machine learning algorithms. (
Enrichment profiling via GO and KEGG revealed a primary association of the core targets with essential BP, such as the phospholipase C-activating G-protein coupled receptor signaling pathway, cellular response to xenobiotic stimulus, positive regulation of the MAPK cascade, and control of tube diameter. Within CC, strong enrichment was identified in structures including the presynaptic membrane, synaptic membrane, gamma-secretase complex, and presynapse. Regarding MF, these targets appeared to be associated with activities such as G-protein coupled amine receptor activity, neurotransmitter receptor activity, carbonic anhydrase activity, and dopamine neurotransmitter receptor activity.
Several key signaling pathways, most notably the calcium, phosphoinositide 3-kinase-protein kinase b (PI3K-Akt), Notch, and Rap1 signaling pathways, were significantly enriched among the core targets, as revealed by KEGG analysis. These results suggest LNT’s potential mechanisms against IVDD, along with its associated BPs and signaling pathways.
Integration of ML and bioinformatics for core therapeutic target screening
In this investigation, three distinct ML approaches—LASSO regression, RF, and XGBoost—were utilized to identify pivotal genes through a structured feature selection process. As shown in Figure 2a, b, LASSO regression utilized the “lambda.min” determined through cross-validation as the optimal regularization parameter, ultimately identifying five core genes: B4GALT1, GGTA1, IL2RA, NMUR2, and STAT3. The RF model assessed gene importance based on the “Mean Decrease Gini” metric, identifying the four genes that contributed most to sample classification, ranked in descending order of importance as SLC6A2, HTR2A, FGF2, and PSENEN (Fig. 2c,d). The ROC curve analysis yielded an AUC value of 0.75 (Fig. 2e), indicating that the model’s ability to discriminate was substantially superior to random classification (AUC = 0.5). Based on feature importance scores, the XGBoost model identified the ADRA1A gene, which exhibited the most significant reduction in loss during decision tree splitting, suggesting its key driver role in phenotype prediction (Fig. 2f). By combining the three methods mentioned above, a total of 10 key genes were identified: B4GALT1, GGTA1, IL2RA, NMUR2, STAT3, SLC6A2, HTR2A, FGF2, PSENEN, and ADRA1A. The 46 overlapping candidate genes were further analyzed using the STRING database to construct a PPI network. By intersecting the PPI-derived and ML-derived gene sets, two core targets, STAT3 and FGF2, were ultimately identified as the most promising candidates for subsequent MD validation (Fig. 2g).
In the GSE15227, GSE70362, and GSE124272 datasets, the AUC values for FGF2 and STAT3 were 0.889 and 0.722, 0.631 and 0.731, and 0.594 and 0.750, respectively. These AUC values indicate that both FGF2 and STAT3 demonstrate favorable discriminative performance across different IVDD datasets (Fig. 3a–c). Based on these core genes, we further constructed a nomogram prediction model to assess the risk of IVDD in individuals (Fig. 3d–f).
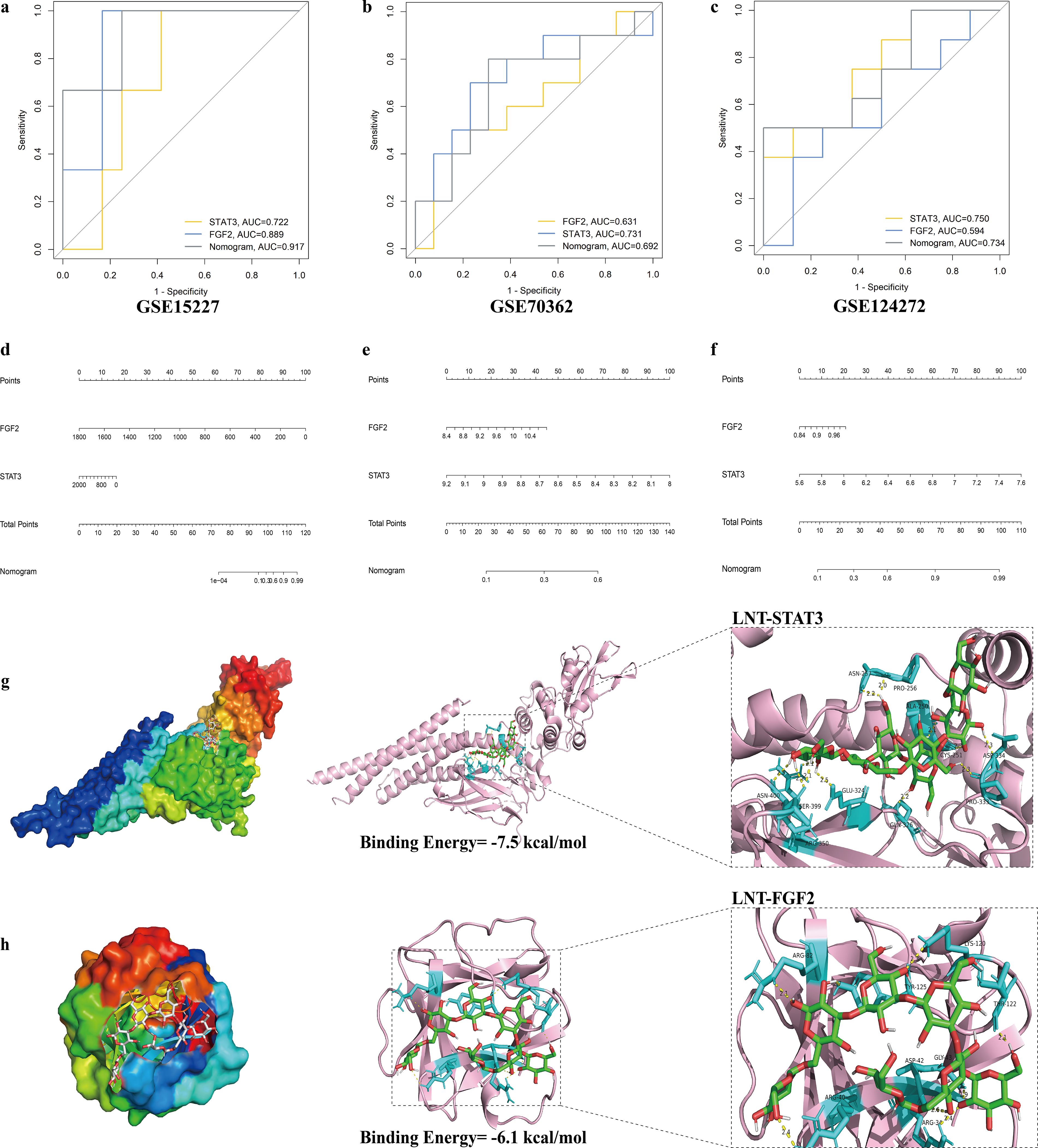
Assessment of LNT–core target interactions using molecular docking and dynamics simulations
The targets STAT3 (PDB ID: 6NJS) and FGF2 (PDB ID: 1BAS) were selected for molecular docking with LNT. The computed minimum binding energies for the LNT–STAT3 and LNT–FGF2 complexes were −7.5 and −6.1 kcal/mol, respectively. These values are significantly below the conventional threshold of −5.0 kcal/mol, indicating strong binding affinity and spontaneous binding capability of LNT toward both targets. The results imply that STAT3 and FGF2 may serve as critical mediators in the regulatory mechanism through which LNT mitigates IVDD. We employed PyMOL to visualize the most stable docking pose (Fig. 3g,h), which delineated the specific binding mode governing the interaction of LNT with its core targets.
To evaluate the dynamic behavior of the LNT complexes, 50 ns MD simulations were performed. The high structural stability of the STAT3–LNT complex throughout the simulation is indicated by its RMSD, which remained stable at an average of 0.45 nm after the system reached equilibrium at ∼10 ns (Fig. 4a). The corresponding RMSD profile for the FGF2–LNT complex is also presented. Residue-level flexibility was analyzed via RMSF, revealing that specific regions, namely, residues 500–1000 of STAT3 and residues 500–750 and 7500–8000 of FGF2, exhibited greater fluctuations, which might correspond to binding sites (Fig. 4b). In contrast, other regions displayed only slight variations, indicating that the overall backbone remained stable. The radius of gyration (Rg) for both complexes exhibited minimal fluctuations, confined to a narrow range of 0.7–0.8 nm throughout the simulation, a finding consistent with a stable and compact folded state (Fig. 4c). The presence of 8–10 stable hydrogen bonds in the STAT3–LNT complex indicates that the ligand forms critical interactions with key residues in the protein’s active pocket. Alternatively, the hydrogen bond count between FGF2 and the ligand varied over time, suggesting that these interactions are dynamic, with bonds constantly forming and breaking (Fig. 5d). The most thermodynamically stable conformational states of both complexes, represented by the blue and purple regions on the Gibbs free energy landscape, correspond to distinct low-energy basins (Fig. 4e,f).
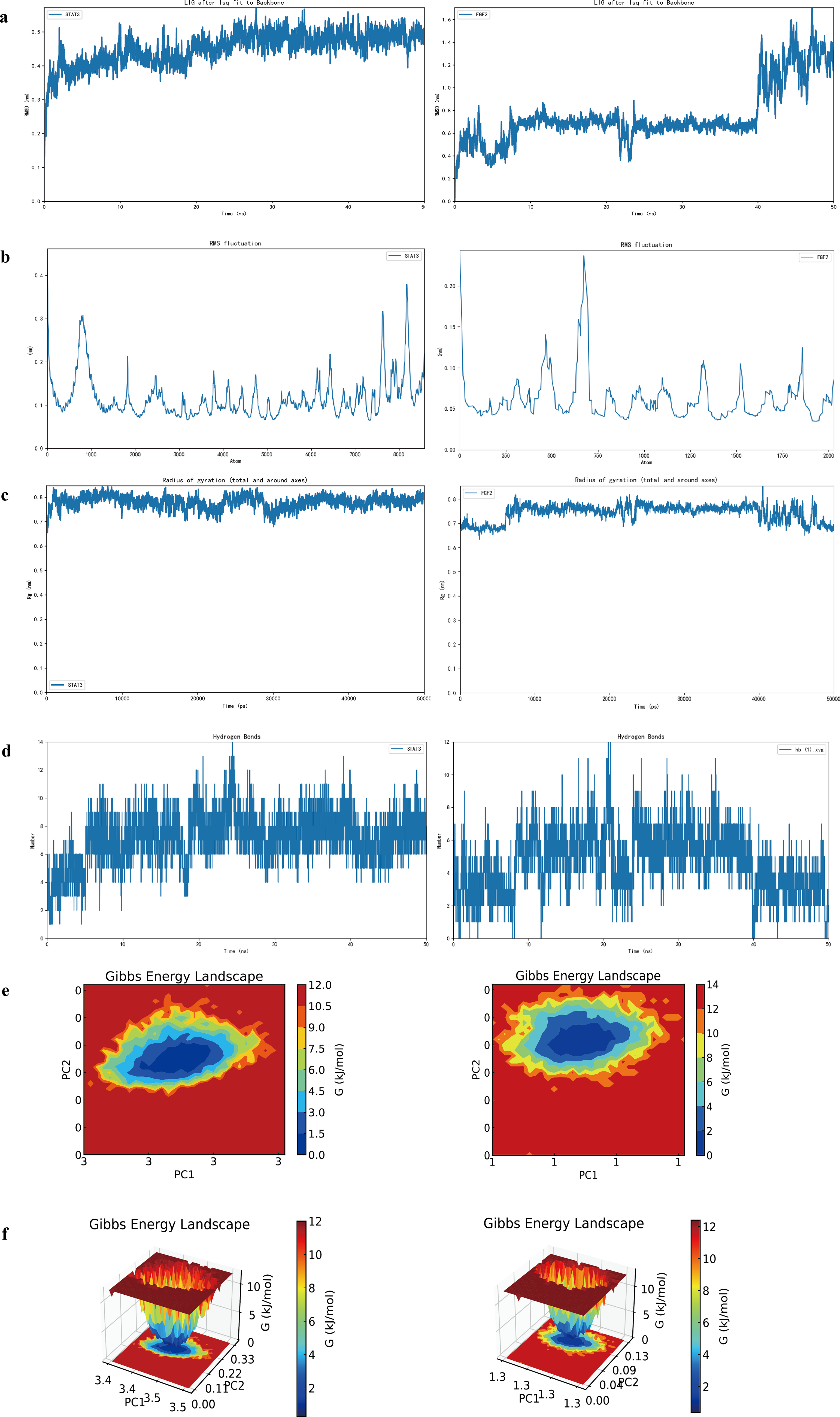
Molecular dynamics simulations of the STAT3–LNT and FGF2–LNT complexes.
In conclusion, the MD simulations showed that the LNT complex maintained relatively stable conformations when interacting with key proteins such as STAT3 and FGF2. Based on these results, LNT appears to engage in stable binding with the identified targets, thereby providing a computational foundation for exploring its potential as a therapeutic agent for IVDD.
Altered immune cell infiltration in IVDD
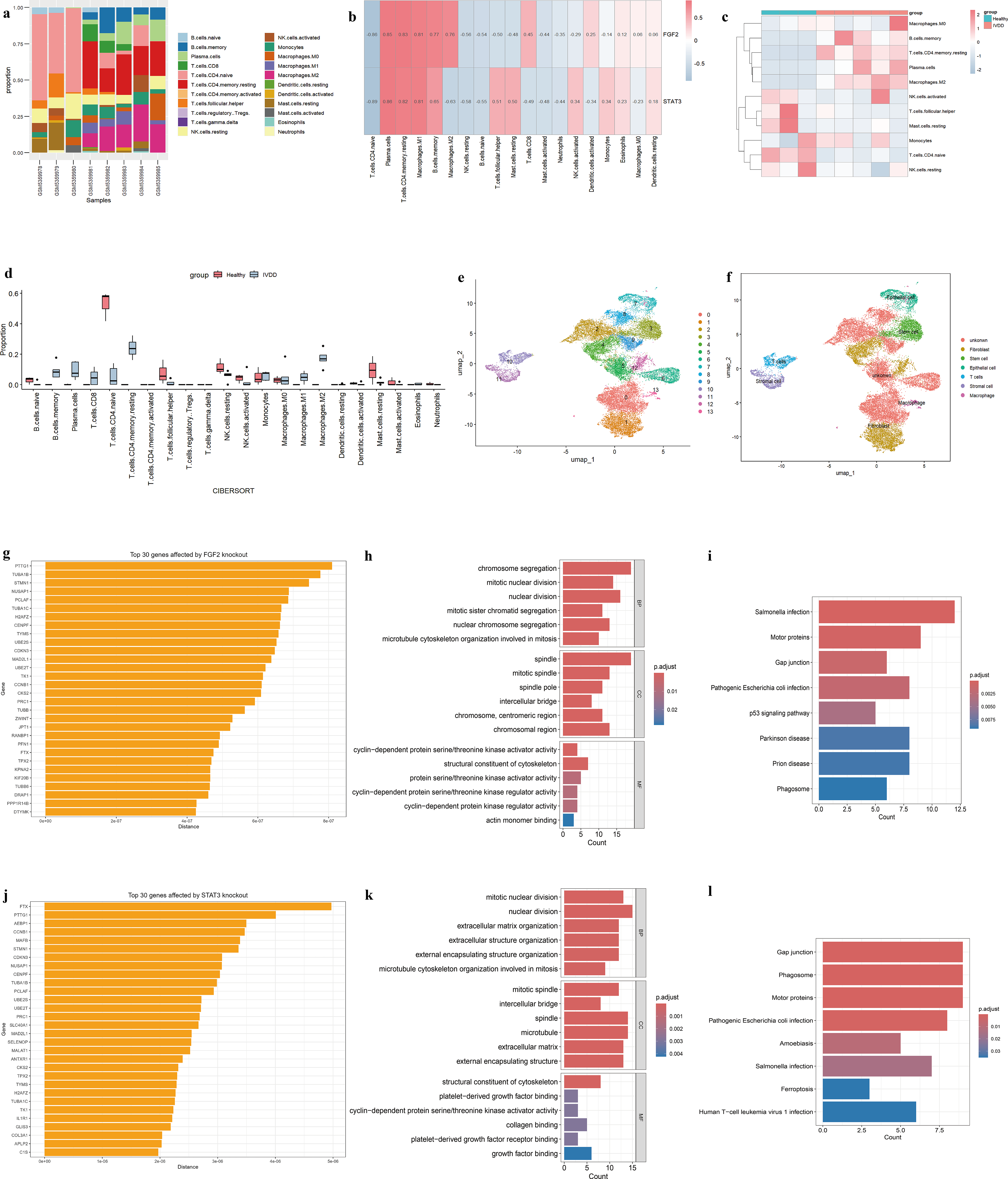
In this study, the CIBERSORT algorithm was applied to quantify the infiltration proportions of 22 immune cell subtypes across all samples. Correlation analysis of immune cells further delineated co-expression and co-occurrence patterns among different immune cell subtypes (Fig. 5a). The relative infiltration levels of distinct immune cell types are shown across samples, with red and blue indicating higher and lower levels, respectively (Fig. 5c). Differential analysis revealed that, compared with IVDD samples, normal samples displayed significantly elevated infiltration proportions of T cells CD4 naive, T cells follicular helper, NK cells resting, and Mast cells resting (Fig. 5d; Supplementary Table S3). In contrast, normal samples exhibited significantly reduced infiltration of B cells memory, plasma cells, T cells CD8, T cells CD4 memory resting, and M1/M2 macrophages, implying their potential involvement in IVDD. Furthermore, we examined the correlation between the expression levels of the two core hub genes, FGF2 and STAT3, and immune cell infiltration. The results indicated that the expression of FGF2 and STAT3 was significantly positively correlated with the infiltration scores of plasma cells, T cells CD4 memory resting, and M1 macrophages, whereas a significant negative correlation was observed with the infiltration score of T cells CD4 naive (Fig. 5b). These findings imply that FGF2 and STAT3 may contribute to reshaping the immune microenvironment during IVDD by modulating the infiltration levels and functional states of specific immune cell subsets.
Identification of downregulated pathways under virtual knockout
To systematically characterize the cellular composition of IVDD tissues and accurately delineate distinct cell types, we performed an in-depth analysis of single-cell RNA sequencing data obtained from IVDD patients. UMAP was employed for dimensionality reduction and visualization, which resolved a total of 13 distinct cell clusters (Fig. 5e). It reflects the presence of significant cellular heterogeneity among the samples. Based on the annotation results, the 13 identified cell clusters were ultimately consolidated and classified into 6 major cell types: fibroblasts, stem cells, epithelial cells, T cells, stromal cells, and macrophages (Fig. 5f; Supplementary Table S4). These results lay the foundation for further investigation into the functional characteristics of different cell subpopulations within the IVDD microenvironment.
The results of the FGF2 virtual knockout analysis revealed that genes such as PTTG1, TUBA1B, and STMN1 exhibited the most significant associated changes (Fig. 5g). Similarly, following the virtual knockout of STAT3, genes such as FTX, PTTG1, AEBP1, CCNB1, MAFB, and STMN1 demonstrated significant correlations (Fig. 5j; Supplementary Table S5). This suggests that the aforementioned genes may play key roles in the regulatory networks mediated by FGF2 and STAT3. Further functional enrichment analysis of the perturbed genes identified during the virtual knockout process revealed that these genes are significantly enriched in BPs such as mitotic cell cycle regulation, cytoskeleton organization, ECM remodeling, the p53 signaling pathway, and ferroptosis-related pathways (Fig. 5h,i, k, l; Supplementary Table S6). Notably, these functional modules are closely associated with the core pathophysiological mechanisms of IVDD, suggesting that FGF2 and STAT3 may participate in the onset and progression of IVDD by regulating the aforementioned key pathways.
Discussion
This study implements a comprehensive bioinformatics approach that integrates network pharmacology, transcriptomic data, and ML algorithms to investigate the potential therapeutic mechanisms of LNT in IVDD. Through LASSO, RF, and XGBoost analyses, STAT3 and FGF2 were identified as core targets mediating the therapeutic effects of LNT. Functional enrichment analysis further revealed that these targets are implicated in multiple signaling pathways associated with inflammation, ECM metabolism, and cellular stress responses. Collectively, these findings provide novel insights into the molecular mechanisms through which LNT may regulate the progression of IVDD.
STAT3, a key transcription factor mediating cytokine signaling, plays a critical role in inflammation, cell proliferation, and immune regulation (Hou and Tian, 2022; Li et al., 2022; Yu et al., 2014). Accumulating evidence suggests that aberrant activation of the JAK2/STAT3 signaling pathway contributes to the progression of IVDD by exacerbating inflammatory responses, promoting ECM degradation, and inducing apoptosis in the NP (Song et al., 2025; Zhu et al., 2025). Furthermore, STAT3 is implicated in diverse processes within the bone microenvironment, such as mesenchymal stem cell differentiation, osteoclast activation, macrophage polarization, and angiogenesis, all of which have the potential to influence intervertebral disc homeostasis (Li et al., 2023; Molnar et al., 2022; Yin et al., 2024). In line with the above findings, our analysis confirmed STAT3 as a core target of LNT in IVDD, indicating that modulating STAT3 signaling likely constitutes a pivotal mechanism underlying the therapeutic action of LNT.
FGF2 is a key member of the FGF family, playing a pivotal role in regulating cell proliferation, differentiation, migration, and survival (Li et al., 2024; Ornitz and Itoh, 2015). In the context of intervertebral disc biology, FGF signaling plays a role in maintaining disc homeostasis through the regulation of ECM metabolism and cellular activity (Li et al., 2024). However, dysregulated FGF signaling has been linked to IVDD. Previous studies indicate that FGF2 promotes the upregulation of matrix-degrading enzymes through FGFR1 signaling, thereby enhancing catabolic activity in disc tissues and facilitating the degradation of ECM components. Furthermore, FGF2 has been demonstrated to counteract the anabolic effects mediated by growth factors, including insulin-like growth factor 1 (IGF-1) and bone morphogenetic protein 7 (BMP7) (Ellman et al., 2008; Loeser et al., 2005). In alignment with the above findings, our differential expression analysis demonstrated a significant upregulation of FGF2 in degenerative disc tissues, suggesting its potential involvement in IVDD progression.
To further investigate the molecular mechanisms by which LNT exerts its therapeutic effects on IVDD, GO and KEGG pathway enrichment analyses were performed. The results revealed that the identified core targets are significantly enriched in multiple signaling pathways, including the PI3K-Akt signaling pathway, calcium signaling pathway, Notch signaling pathway, and Rap1 signaling pathway. Among these pathways, the PI3K/Akt signaling pathway is widely recognized for its protective role in intervertebral disc cells. Its activation facilitates ECM synthesis, suppresses apoptosis, promotes cell proliferation, regulates autophagy, and alleviates oxidative stress damage (Ouyang et al., 2017). Previous studies have further indicated that inflammatory cytokines, including tumor necrosis factor-alpha (TNF-α), are highly expressed in the NP of IVDD patients and are capable of activating both the PI3K/Akt and nuclear factor kappa-light-chain-enhancer of activated b-cells (NF-κB) pathways, thereby exacerbating IVDD (Liao et al., 2022).
The Notch signaling pathway is another critical regulator of cell differentiation and tissue homeostasis (Zieba et al., 2020). Within the microenvironment of the intervertebral disc, inflammatory stimuli can activate Notch signaling, which subsequently alters the equilibrium between anabolic and catabolic processes in the disc tissue (Wang et al., 2013; Zheng et al., 2018). Interestingly, the effect of Notch signaling appears to be cell-type dependent. In AF cells, Notch activation suppresses the expression of collagen type II and aggrecan while concurrently promoting the expression of catabolic factors, thereby facilitating ECM degradation (Liu et al., 2023; Peng et al., 2021; Zheng et al., 2018).
Rap1 signaling is closely linked to integrin-mediated cell adhesion and ECM interactions. Dysregulated Rap1 signaling can compromise integrin signaling, resulting in abnormal ECM remodeling. In addition, Rap1 is thought to participate in angiogenesis through the regulation of VEGFR2 activation (Lakshmikanthan et al., 2011). While the normal intervertebral disc is an avascular tissue, pathological angiogenesis is commonly observed during disc degeneration and is closely linked to inflammation and tissue fibrosis (Kawakubo et al., 2020). Therefore, aberrant Rap1 signaling may contribute to the progression of IVDD through multiple mechanisms.
To further validate the above findings, molecular docking was performed. The results demonstrated significant binding affinity between LNT and two core targets—STAT3 and FGF2, indicating that LNT can spontaneously and stably interact with these target proteins. In the MD simulations, the STAT3–LNT complex exhibited stable RMSD, minimal fluctuations in Rg, low RMSF in key regions, and a high proportion of stable hydrogen bonds, collectively indicating a conformationally stable simulation system. The structural and dynamic characteristics of the protein complex align with functional expectations. In contrast, the RMSD of the FGF2–LNT complex exhibited a sharp rise in the later phases of the simulation, which was accompanied by the detachment of the ligand molecule. This suggests that weaker interactions—such as hydrogen bonds and hydrophobic effects—between the ligand and the binding pocket of FGF2 may have led to abrupt detachment from the binding site, resulting in increased overall conformational deviation. These findings provide dynamic insights for a deeper understanding of the MFs of STAT3 and FGF2, the mechanisms of ligand binding, and guidance for subsequent ligand optimization.
To further investigate the regulatory roles of the identified core genes, virtual knockout analyses were conducted. The virtual knockout of STAT3 and FGF2 revealed multiple perturbed genes, indicating that these genes likely function as downstream targets modulated by the core regulatory network. For example, genes including STMN1 and PTTG1 exhibited strong associations with FGF2 knockout. Prior research has reported a significant upregulation of STMN1 in degenerative NP tissues, implicating it in IVDD progression (Li et al., 2025). Similarly, genes such as FTX, AEBP1, CCNB1, and MAFB showed strong associations with STAT3 knockout. It is noteworthy that AEBP1 has been identified as one of the most significantly enriched hub genes in degenerative intervertebral disc tissues (Dube et al., 2025).
Functional enrichment analysis of these perturbed genes revealed significant enrichment in pathways associated with inflammation, apoptosis, oxidative stress, and cell cycle regulation. These BPs are broadly considered key mechanisms in the pathogenesis of IVDD. Among the enriched pathways, the p53 signaling pathway plays a vital role in maintaining intervertebral disc homeostasis. Activation of p53 can induce apoptosis and cellular senescence in disc cells, leading to a decline in functional cell populations within the disc tissue (Yin et al., 2021; Zhang et al., 2021). In addition, p53 may contribute to the development of a senescence-associated secretory phenotype, thereby disrupting the equilibrium between anabolic and catabolic processes in the intervertebral disc microenvironment (Wang et al., 2023). The enrichment of perturbed genes within the p53 pathway suggests that disruption of the core targets may influence disease progression by modulating p53-mediated stress responses.
Ferroptosis is an iron-dependent form of regulated cell death characterized by lipid peroxidation and redox imbalance (Imam et al., 2017). It has recently been implicated in the development of IVDD. Accumulating evidence suggests that ferroptosis promotes oxidative damage and inflammatory responses in degenerative intervertebral disc tissues (Sun et al., 2025; Wang et al., 2022; Yang et al., 2023; Yu et al., 2022; Zhang et al., 2021; Zhu et al., 2023). The enrichment of ferroptosis-related pathways among the perturbed genes indicates that the identified core targets may regulate the progression of IVDD by influencing molecular events associated with ferroptosis.
Taken together, these findings indicate that LNT likely exerts its therapeutic effects on IVDD by targeting key genes, notably STAT3 and FGF2. Through modulation of these targets, LNT can impact multiple downstream signaling pathways, such as PI3K-Akt, Notch, Rap1, and p53 signaling, as well as ferroptosis-associated pathways. Collectively, these pathways govern essential cellular processes including inflammation, apoptosis, oxidative stress, and ECM metabolism, ultimately shaping the progression of IVDD.
In external dataset validation, although FGF2 achieved an AUC of only 0.594 in the GSE124272 cohort, it was consistently identified as a consensus target by both PPI network analysis and ML models, suggesting its potential biological relevance. The relatively low AUC value may be attributed to cohort heterogeneity and dataset quality.
Although the limited size of the discovery cohort (3 controls and 5 IVDD samples) may have compromised the robustness of DEG analysis and ML outputs, this concern was partially mitigated by subsequent validation using multiple external datasets. This integrated approach overcomes the limitations of single-omics data and significantly enhances both the accuracy and interpretability of target prediction. It is well recognized that validating the outcomes derived from network-based computational methods is essential, requiring well-planned experimental approaches to confirm the reliability of the findings. To translate these basic findings into clinical applications, the imperative next step is the rigorous experimental validation of LNT in carefully designed in vitro and in vivo models. A major focus of this translational research should be assessing whether LNT can serve as an adjunct therapy to slow IVDD development and mitigate associated low back pain, thus transforming predictive data into validated evidence.
Conclusion
This study employed an integrated bioinformatics and computational approach, encompassing network pharmacology, ML, molecular docking, MD simulations, immune infiltration analysis, and virtual knockout, to systematically investigate the potential molecular mechanisms underlying LNT’s intervention in IVDD. Our findings demonstrate that LNT interacts with two core targets, STAT3 and FGF2. These targets are implicated in multiple critical BPs, including the phospholipase C-activated G-protein-coupled receptor signaling pathway, response to xenobiotic stimuli, positive regulation of the MAPK cascade, and regulation of vascular lumen diameter. Moreover, they exhibit significant enrichment in key signaling pathways such as the calcium signaling pathway, PI3K-Akt signaling pathway, Notch signaling pathway, and Rap1 signaling pathway. These observations suggest that LNT likely exerts its therapeutic effects on IVDD by modulating these processes and pathways.
Molecular docking results indicate that the binding energies of LNT with the core targets STAT3 and FGF2 are both below −5 kcal/mol, reflecting favorable and stable binding interactions. Subsequent MD simulations further confirm the robust binding affinity and complex stability between LNT and these targets. In addition, virtual knockout analysis of the core genes provides further mechanistic insight, revealing downstream signaling pathways in IVDD that may be modulated under LNT regulation.
Collectively, this work not only elucidates potential molecular mechanisms by which LNT alleviates IVDD but also broadens the application of integrated methodologies—including network pharmacology, molecular docking, and MD simulations—in probing the mechanisms of active components in traditional Chinese medicine. This study offers novel perspectives and robust research tools for understanding the pharmacological effects of LNT.
Authors’ Contributions
H.C.: Writing—original draft, supervision, project administration, and conceptualization; Y.C.: Writing—original draft and funding acquisition; H.L.: Supervision, project administration, funding acquisition, and conceptualization; T.Z.: Supervision and project administration.
Footnotes
Availability of Data and Materials
The data that support the findings of this study are available in the Gene Expression Omnibus database with the accession number GSE176205 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE176205), GSE15227 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE15227), GSE70362 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE70362), GSE124272 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE124272) and GSE230808 (![]() ). These data were derived from the above-mentioned public domain resource.
). These data were derived from the above-mentioned public domain resource.
Author Disclosure Statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.
Funding Information
This work was supported by