
Abstract
Chronic low-grade inflammation has been identified as the etiology of disease across the life span; however, the underlying genetic mechanism are poorly understood, and no genetic association study is performed. Here, we quantified chronic low-grade inflammation using the low-grade inflammation score (INFLA-score) and performed a genome-wide association study in a large European cohort of 217,984 individuals, involving 6,134,151 single nucleotide polymorphisms (SNPs). We identified 20,182 SNPs at 194 loci reaching genome-wide significance (p < 5 × 10−8), with the lead SNP rs429358 in apolipoprotein E (APOE) showing an extremely low p-value of 8.69 × 10−166. Gene analysis found 470 genes with p < 2.70 × 10−6 (0.05/18,519), among which phosphodiesterase 4B was most significant (p = 1.67 × 10−22). Among those, 15 SNPs were first reported, including GS1-259H13.10, AC068533.7, AL355490.1, and so on. Tissue expression analysis revealed significant links between genetic variants and chronic low-grade inflammation in spleen and whole blood. Our findings identify variants linked to chronic low-grade inflammation and related inflammatory and cellular regulation traits, and suggest a close association between chronic low-grade inflammation and the spleen.
Introduction
Systemic chronic inflammation (SCI) refers to a prolonged inflammatory response triggered by various stimuli, injury, or dysregulated acute inflammation (Nasef et al., 2017). Transitioning from a transient to a persistent inflammatory response can disrupt immune tolerance and result in significant changes in tissue and organ functioning, as well as cellular physiology. This shift in inflammatory dynamics poses an increased risk for the development of various inflammatory diseases (Gaikwad et al., 2025), such as the risk of NAFLD (Xue et al., 2025). It is also strongly linked to oxidative stress (Sater et al., 2025). Notably, chronic inflammatory diseases have emerged as significant contributors to morbidity and mortality worldwide, accounting for >50% of total deaths (Vos et al., 2020). These inflammation-related conditions encompass a broad range of diseases, including ischemic heart disease, stroke, cancer, diabetes mellitus, chronic kidney disease, autoimmune disorders, and neurodegenerative disorders (Kadatane et al., 2023; Khanna et al., 2022; Pounis et al., 2016; van de Vyver, 2023). However, the potential molecular pathways linking SCI with chronic diseases are poorly understood.
While the associations between SCI and disease risk and mortality are well documented (Cifuentes et al., 2025; Pounis et al., 2016), the current lack of standardized biomarkers hinders the identification of health-damaging chronic inflammation. The use of a low-grade inflammation score (INFLA-score) has been previously employed to assess the potential synergistic effects of multiple inflammatory biomarkers, including C-reactive protein (CRP) levels, white blood cell (WBC) counts, platelet (PLT) counts, and neutrophil-to-lymphocyte ratios (NLR) (Cheng et al., 2024), are inexpensive biomarkers of systemic inflammation (SI) (Tang et al., 2025). In the Moli–Sani study, higher dietary polyphenol intake was significantly associated with lower INFLA-scores, reflecting reduced levels of low-grade inflammation. This finding underscores the potential anti-inflammatory benefits of polyphenol-rich diets, which may contribute to a lower risk of inflammation-related diseases (Liu and Gu, 2024). Higher INFLA-scores have also been associated with increased risk of cardiometabolic diseases, elevated diabetes risk mediated by body mass index (BMI) (Cheng et al., 2024; Li et al., 2024), and poor functional outcomes in acute ischemic stroke (Zhou et al., 2023). Furthermore, elevated INFLA-scores have been correlated with poorer functional outcomes in patients suffering from acute ischemic stroke. Higher scores are associated with greater stroke severity and early neurological deterioration, suggesting that the INFLA-score could serve as a valuable prognostic tool in acute stroke management (Bao et al., 2024; Liu et al., 2022). Additionally, concurrent changes in inflammatory biomarkers observed among patients suggest that accounting for these synergistic effects may provide a more holistic understanding of the inflammatory state.
To investigate the genetic variants linked to SCI, we conducted a genome-wide association study (GWAS) utilizing the UK Biobank cohort. Subsequently, functional analyses were performed on the identified loci to gain insight into the underlying biological processes that may regulate chronic inflammation.
Materials and Methods
UK biobank cohort
The recruitment of the UK Biobank cohort took place between 2006 and 2010, encompassing a population of >500,000 individuals aged between 37 and 73 years across Great Britain (England, Wales, and Scotland) (Tan and Timpson, 2022). Ethical approval was obtained from the UK Biobank, and all participants provided informed consent. Details of the UK Biobank resource can be found at www.ukbiobank.ac.uk. The UK Biobank received ethical approval from the National Health Service National Research Ethics Service (reference 11/NW/0382). The current analyses were conducted under the approved UK Biobank (data application number 65711).
Genotyping and quality control
The UK Biobank provides ∼96 million variants, which have been imputed using the UK10K haplotype, 1000 Genomes Phase 3, and Haplotype Reference Consortium reference panels. A GWAS analyzing SCI was conducted utilizing data from the v3 release of the UK Biobank. Quality control metrics were established and employed by the UK Biobank to filter DNA variants (Lucius, 2023). The genomic position used throughout this study was the human genome assembly GRCh37 (hg19) from the Genome Reference Consortium.
In this study, we used PLINK2.0 (https://www.cog-genomics.org/plink/2.0) as the main GWAS software and removed single nucleotide polymorphisms (SNPs) with an imputation score <0.8, a minor allele frequency <0.01, a genotype missingness >0.05, or a failed Hardy–Weinberg test p < 1 × 10−6.
The quality control criteria for the participants included removing (1) those who were “nonwhite-British” based on self-identification and a principal component analysis of the genotypes, (2) individuals who carried sex chromosome configurations that were not either XX or XY, (3) outliers in heterozygosity and missing rates, (4) those with a sample missing rate > 0.05, (5) individuals whose genetic sex differed from the self-reported value, (6) individuals with no kinship found, and (7) participants who withdrew. A sample of 276,117 individuals and 6,134,154 SNPs was left for the GWAS.
Phenotypic information on the INFLA-score
Although various indicators of inflammation have been proposed in the field of SCI, no gold standard biomarker exists (Xue and Zhao, 2025). The INFLA-score was calculated from various biomarkers, including CRP (Field ID:30710, CRP), WBC (Field ID:30000, WBC [leukocyte] count), PLT (Field ID:30080, PLT count), and NLR (Field ID:30200, Neutrophil percentage; Field ID:30180, Lymphocyte percentage), from the Moli–Sani study (Khanna et al., 2022).
Table 1 shows the scoring system utilized for constructing the INFLA-score. Participants who were receiving immune-modulating drug treatment, were diagnosed with autoimmune-related diseases or disorders, or had blood diseases and hematologic neoplasms were excluded from the study (Supplementary Table S1). Additionally, for each biomarker, individuals with extreme values beyond ±3 standard deviations from the mean were excluded. The value of each biomarker was divided into 10 quantiles, where the highest deciles (7–10) were assigned scores ranging from 1 to 4. Conversely, the lowest deciles (1–4) received negative scores ranging from −4 to −1, while deciles 5 or 6 were assigned zero points. The values of the four biomarkers were subsequently aggregated to derive the INFLA-score, which represents the degree of SCI on a scale from −16 to 16. A higher INFLA-score indicates more pronounced SCI.
The Scoring System Used for the Construction of the Low-Grade Inflammation Score

The INFLA-score, which is a summation of the four components, ranged from −16 to 16.
The biomarker levels were divided into 10 quantiles (Q1–Q10), and each quantile was assigned a score using the established scoring system.
CRP, C-reactive protein; NLR, neutrophil-to-lymphocyte ratio; PLT, platelets; WBC, white blood cells.
Genome-wide association study
To identify genetic variants associated with SCI, we used PLINK2.0 to construct a linear regression model assuming an additive effect adjusted for age at recruitment, sex, genotyping array, BMI, smoking status, and the top 20 principal components of genetic ancestry. Linkage disequilibrium score (LDSC) regression was used to estimate the genetic correlation between the GWAS results and SCI. Additionally, the LDSC intercept was examined to assess residual confounding in the meta-analysis (Bulik-Sullivan et al., 2015; Watanabe et al., 2017).
Independent loci were independently analyzed in the meta-analysis using the default settings in FUMA (Functional Mapping and Annotation of Genome-Wide Association Studies) (Groenewoud et al., 2022). Independent lead-type SNPs were identified based on their significance (p < 5e-8) and independence (r2 < 0.6). Among these independent SNPs, lead SNPs were further defined as those with low linkage disequilibrium (LD) at r2 < 0.1. Loci were defined by combining lead SNPs within a 500 kb window and including all SNPs in LD (r2 ≥ 0.6) with at least one of the independent SNPs. Quantile–quantile (Q–Q) and Manhattan plots were created using CMplot.
Gene analysis, gene set analysis, and tissue expression analysis
The FUMA web application was utilized to apply SNP functional annotations. Within FUMA, the Multimarker Analysis of GenoMic Annotation (MAGMA) tool was used to compute gene-based p values (gene analysis) and gene set p values (gene set analysis) using the provided GWAS summary statistics (Said et al., 2022). In the gene analysis, SNP-to-gene mapping was performed specifically for protein-coding genes when the SNPs were located within them, resulting in the computation of gene-based p values. In the gene set analysis, gene-based p values were calculated for 4,728 curated gene sets (including canonical pathways) and for 6,166 GO terms obtained from MsigDB v5.2. For both analyses, the default MAGMA settings were used, utilizing the SNPwise model for gene analysis and the competitive model for gene set analysis. To account for multiple testing, the Bonferroni correction was applied for gene analysis, while the false discovery rate correction was used for gene set analysis (Said et al., 2022).
In the tissue expression analysis by GTEx integrated in the FUMA, average gene expression per tissue type was used as a gene covariate to test for a positive relationship between gene expression in a specific tissue type and genetic associations. Two types of tissue analysis were performed. One used 30 general tissue types, and the other used 53 specific tissue types.
RESULTS
GWAS results
The characteristics of study participants are summarized in Table 2. Male participants, who were generally younger, had lower BMI, and exhibited a lower frequency of smoking, were found to have lower levels of chronic inflammation. This suggests that factors such as age, BMI, and smoking habits may play a role in modulating chronic inflammation within this population. The relationship between these variables and inflammation could reflect underlying biological or lifestyle factors that contribute to the inflammatory profiles observed in the study group. After quality control, 217,984 participants of European descent and 6,134,154 SNPs remained for subsequent genetic association analysis (Methods), which identified 20,182 SNPs associated with an INFLA-score (at a genome-wide significance of p < 5 × 10−8). (Fig. 1a, Supplementary Table S2). We compared the observed p values to the distribution of p values expected by chance in a Q–Q plot (Fig. 1b). The blue dots deviate from the red line, indicating that the observed association signals are significantly stronger than the signals that would be expected by chance.
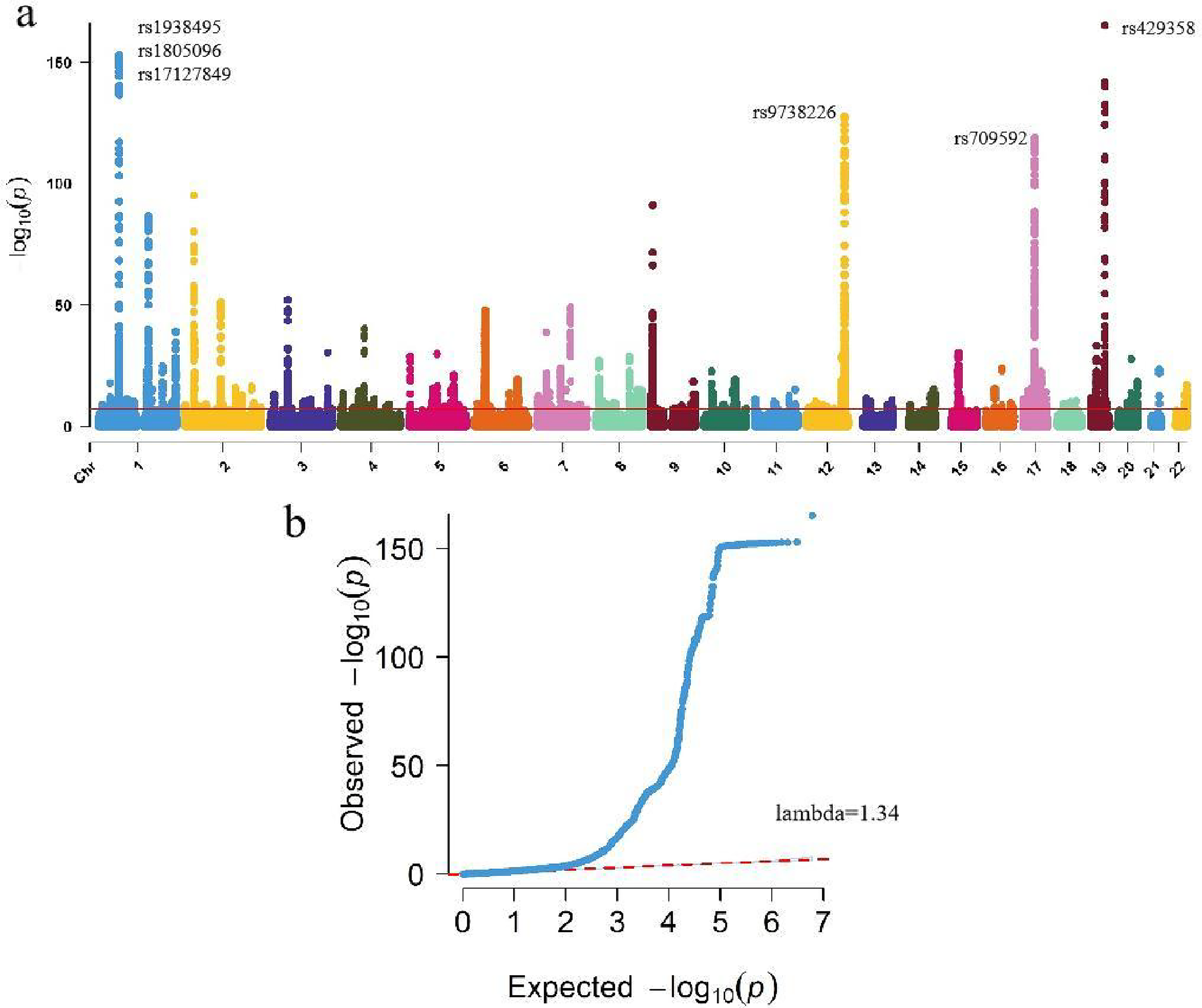
Genome-wide association analysis of low-grade inflammation.
Study Demographics
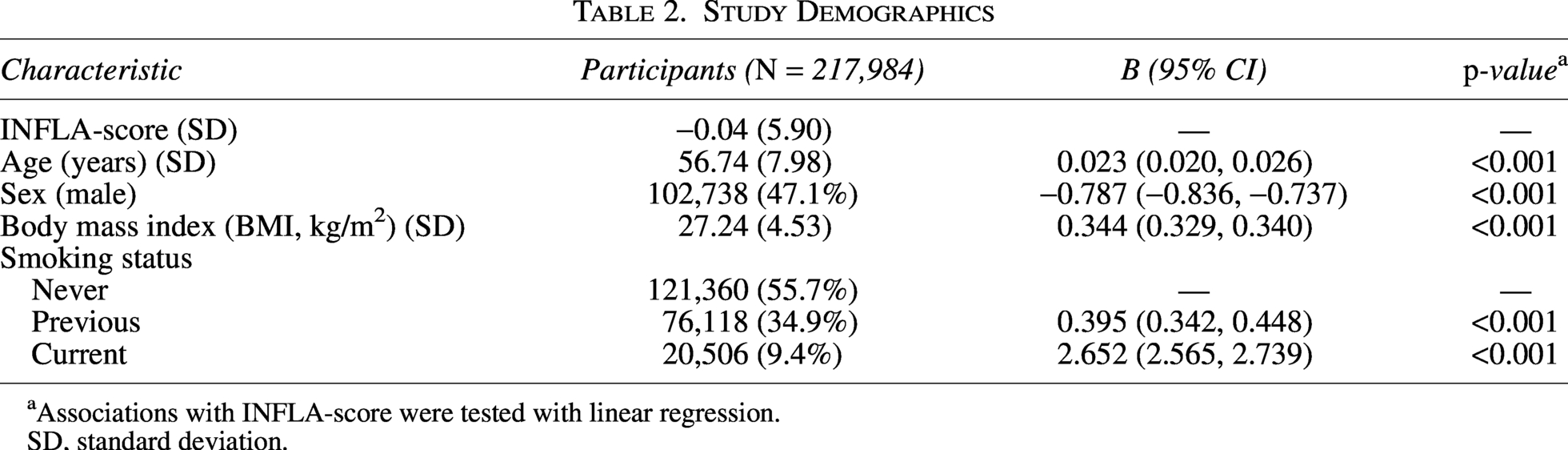
Associations with INFLA-score were tested with linear regression.
SD, standard deviation.
Of these, we mapped 330 lead SNPs, 872 independent significant SNPs and 1,116 mapped genes with p < 5 × 10−8 from 194 genomic risk loci (Supplementary Tables S3 and S4) by using the FUMA platform. Genomic risk loci defined by independent lead SNPs and maximum distance between their LD block. We observed the strongest association for rs429358 at apolipoprotein E (APOE), TOMM40, APOC1 and NKPD1 (p = 8.69 × 10−166) (Fig. 2a), followed by rs1938495 at the LEPR (p = 1.40 × 10−153) (Fig. 2b). The LDSC regression intercept term from the GWAS output was 1.1097 (SE 0.0183), and approximately 16.3% of the genomic inflation (λ = 1.344) was due to polygenic signals rather than sources of confounding factors, including population stratification and cryptic relatedness.
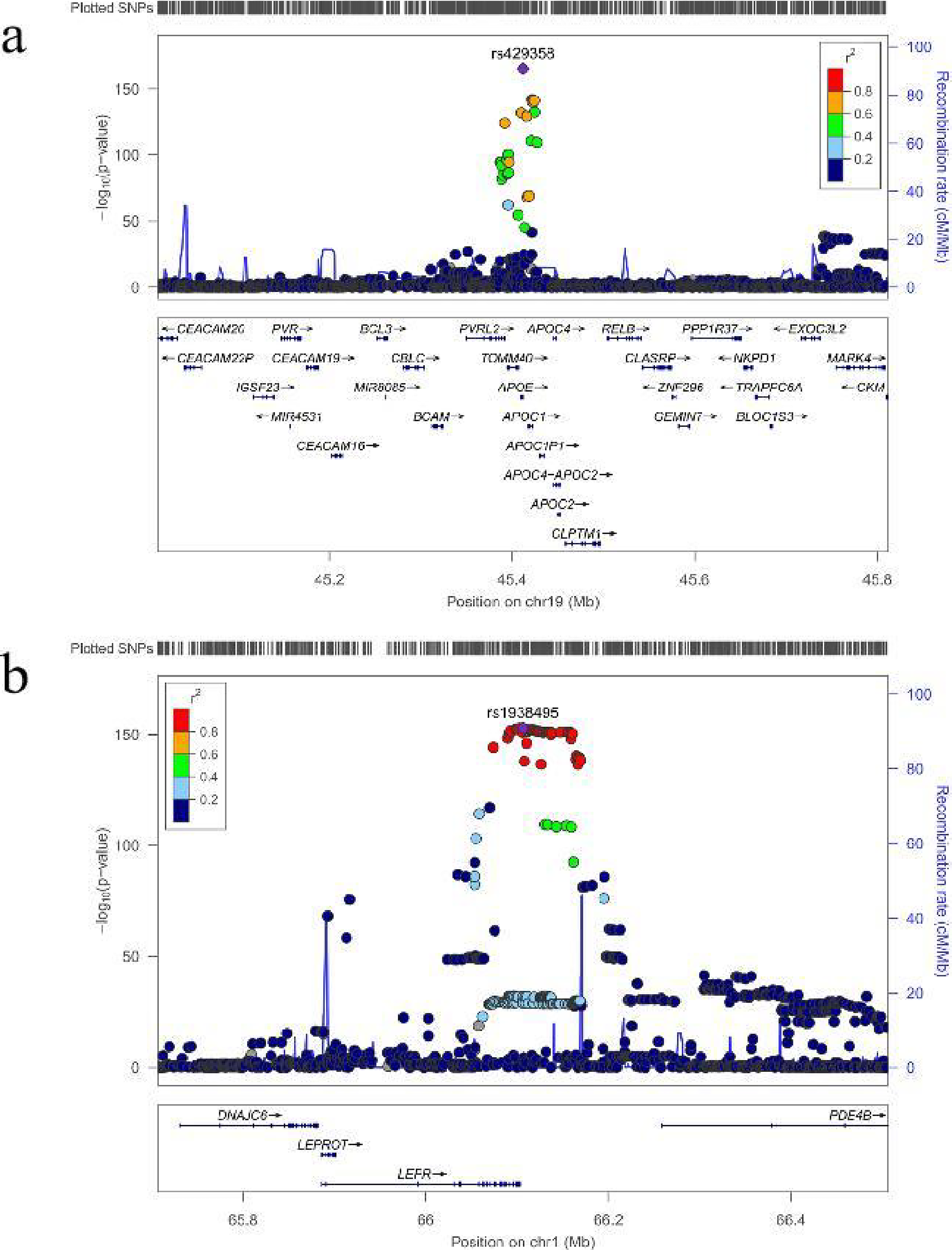
LocusZoom plots of the top associated genomic risk loci.
Gene analysis, gene set analysis and tissue expression analysis by FUMA
The gene analysis using MAGMA integrated in FUMA resulted in the mapping of all SNPs to 18,519 protein-coding genes, provided that the SNPs were located within the genes. Notably, phosphodiesterase 4B (PDE4B) exhibited the most significant association, with a p-value of 1.67 × 10−22, followed by death domain-associated protein (DAXX) (p = 5.55 × 10−17), nuclear receptor binding protein 1 (NRBP1) (p = 1.11 × 10−16), BCL2 antagonist/killer 1 (BAK1) (p = 2.22 × 10−16), and interleukin 36 gamma (IL36G) (p = 3.89 × 10−16). Supplementary Table S5 includes all genes (n = 470) with a p-value less than the threshold of 2.70 × 10−6 (0.05/18,519).
In the gene set analysis by MAGMA integrated in FUMA, a total of 17,005 gene sets were tested. The gene set related to cellular response to biotic stimuli had the lowest p-value (9.23 × 10−9), followed by the gene sets related to positive regulation of RNA metabolism (p = 1.32 × 10−8), transcriptional regulation of granulopoiesis (p = 1.58 × 10−8), regulation of immune system processes (p = 1.59 × 10−8), and regulation of immune response signaling pathways (p = 4.41 × 10−8). All the gene sets (n = 34) with p < 2.94 × 10−6 (0.05/17,005) are included in Supplementary Table S6.
According to the results of the expression analysis of the 30 general tissue types, the whole-blood tissue exhibited the most significant difference (p = 1.40 × 10−9), followed by the spleen tissue (p = 5.79 × 10−9) (Fig. 3; Supplementary Table S7). Among the 53 specific tissue types, tissues from the spleen had the lowest p-value (1.15 × 10−11), and whole blood and lung tissue also had p values of 1.25 × 10−11 and 1.29 × 10−8, respectively (Fig. 3; Supplementary Table S8).
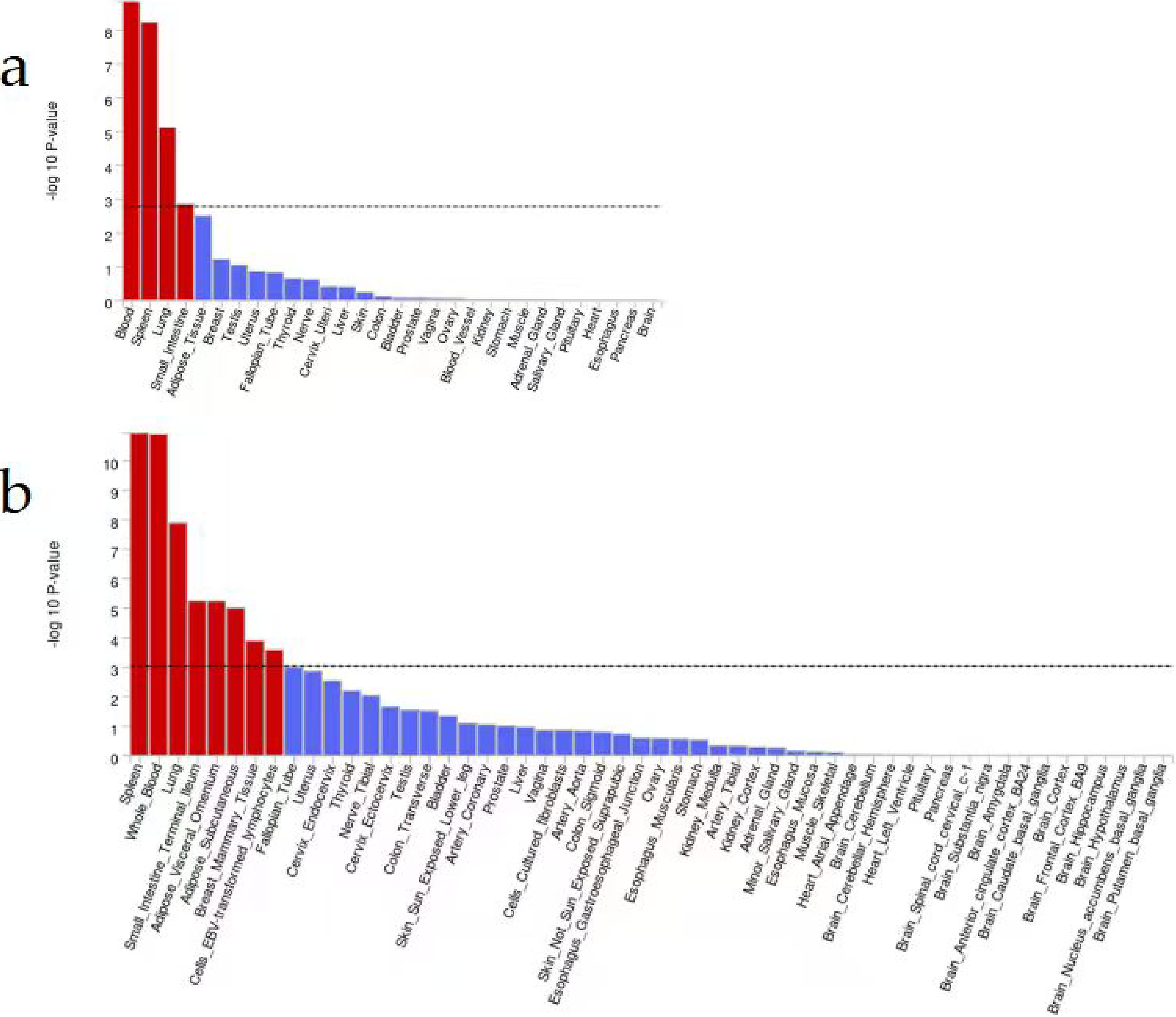
Tissue expression profiles across different tissue types based on GTEx in the FUMA cohort.
Discussion
We scored SCI using the UK Biobank resource as a proxy for the inflammatory state of the organism and performed a GWAS. Our analysis of 217,984 individuals revealed that 166 loci were significantly associated with SCI. The variance explained by these independent variants within the GWAS loci was 16.3%. Evidence from tissue expression analysis showed that the spleen was implicated in SCI at the genetic level.
CRP has been extensively utilized as a biomarker for SI in genetic studies, research shows that general obesity, but not abdominal obesity, is associated with worsening depressive symptoms and incident depression, which can be partly explained by the systemic inflammatory response defined by CRP levels (Guo et al., 2023). Koskeridis et al. and Said et al. highlighted the pleiotropic genetic basis of CRP levels, revealing overlapping pathways between CRP and metabolic, cardiovascular, and immune-related conditions (Koskeridis et al., 2022; Royall, 2022). While the present study does not directly measure CRP, the INFLA-score, a composite phenotype encompassing multiple traits associated with SCI, exhibits considerable genetic overlap with loci identified in the CRP GWAS. Several critical loci previously identified in CRP studies were replicated in this analysis. Both referenced papers underscore the role of LEPR in regulating inflammatory responses, which is supported by the identification of rs1938495 as a significant signal in this study. Similarly, the centrality of APOE in influencing CRP levels, highlighted by Said et al., is consistent with the observed association of rs429358 within the APOE region (Chaloemtoem et al., 2024). In addition to these established loci, the present study identified novel associations, including PDE4B and IL36G, which are implicated in immune system regulation and cytokine production. These findings expand the genetic landscape of chronic inflammation and suggest previously uncharacterized biological pathways. Tissue expression analysis revealed significant enrichment of associated loci in immune-related tissues, such as whole blood and the spleen, emphasizing the systemic nature of SCI. While Koskeridis et al. and Said et al. identified the liver as a key site for CRP synthesis, these results indicate a broader involvement of immune-specific tissues in inflammation processes. Gene set enrichment analysis conducted via MAGMA highlighted pathways such as “cellular response to biotic stimuli” and “regulation of immune response signaling,” which is consistent with the CRP-related pathways identified in prior studies (Koskeridis et al., 2022; Royall, 2022). Furthermore, the prioritization of pathways related to RNA metabolism and granulopoiesis reflects broader biological mechanisms underlying inflammation, providing a more comprehensive genomic interpretation than does CRP-focused analyses.
In our GWAS, we identified 166 loci associated with SCI. Notably, one locus in the APOE gene, the APOC1 gene (rs429358), exhibited a significantly low p-value of 8.69 × 10−166. The APOE4 allele of the APOE gene is a widely recognized genetic risk factor for Alzheimer’s disease (AD), and its association with decreased hippocampal volume and elevated amyloid-β plaque levels in the brain (Chaloemtoem et al., 2024; Koskeridis et al., 2022) when combined with chronic low-grade peripheral inflammation, contributes to earlier onset and increased morbidity of AD (Parhizkar and Holtzman, 2022). The APOC1 gene encodes a member of the apolipoprotein C1 family that is primarily expressed in the liver. Its activation occurs during the differentiation of monocytes into macrophages. This protein plays a pivotal role in the metabolism of high-density lipoprotein (HDL) and very low-density lipoprotein (LDL) (Rouland et al., 2022). Additionally, APOC1 inhibits hepatic lipase (Kinnunen and Ehnolm, 1976), which is responsible for triglyceride (TG) hydrolysis in cholesterol-rich lipoproteins, including HDL. Moreover, APOC1 is implicated in various facets of lipoprotein metabolism, as well as in the modulation of inflammatory responses and the biology of specific cellular components within the vascular wall (Rouland et al., 2022).
The LEPR gene (rs1938495) had a significant p-value of 1.40 × 10−153. In agreement with these findings, the gene analysis highlighted signaling by leptin as an enriched gene set. The LEPR gene, a member of the gp130 family of cytokine receptors, is known for its ability to activate cytosolic STAT (Signal Transducers and Activators of Transcription) proteins and stimulate gene transcription. The rs7799039 variant of the LEPR gene has been demonstrated to be associated with improved LDL and TG levels following weight loss (Primo et al., 2021). Another study revealed a positive association between the LEPR G3057A mutation and TG and LDL levels but a negative association with HDL levels in Chinese diabetic patients (Zhang et al., 2018). These findings suggest that genetic variations within the LEPR gene may contribute to disturbances in lipoprotein metabolism and the development of metabolic disorders such as obesity and type 2 diabetes.
The results of the gene analysis highlight several protein-coding genes with significant associations, notably PDE4B, DAXX, NRBP1, BAK1, and IL36G. These findings provide a valuable foundation for exploring their potential roles in SCI and related biological pathways. PDE4B, a prominent member of the PDE4 family, plays a critical role in regulating cyclic adenosine monophosphate (cAMP) signaling pathways, which are essential for modulating inflammatory responses. By specifically hydrolyzing cAMP, a second messenger recognized for its potent anti-inflammatory effects, PDE4B effectively diminishes the inhibitory impact of cAMP on inflammatory pathways. In a word, it regulates chronic inflammation via the cAMP-NF-κB pathway, representing a novel potential anti-inflammatory drug target distinct from CRP (Chinn et al., 2022; Su et al., 2022; Tavares et al., 2020). DAXX is a multifunctional protein with diverse roles in histone chaperoning, transcriptional regulation, and apoptotic signaling. As a histone H3.3 chaperone, DAXX facilitates the deposition of H3.3 into heterochromatin by interacting with ATRX, thereby contributing to chromatin remodeling and genomic stability. This function is essential for preventing telomere dysfunction and maintaining chromosomal integrity. In addition to its role in chromatin regulation, DAXX modulates transcriptional activity through interactions with SUMOylated proteins and transcription factors, influencing pathways involved in apoptosis and DNA repair (Bogolyubova and Bogolyubov, 2021). Furthermore, DAXX plays a critical role in cellular stress responses by regulating inflammatory pathways. For example, the FAS-DAXX-ASK1 pathway links DAXX to apoptosis triggered by inflammatory stimuli, where ASK1 activation enhances MAPK signaling, ultimately inducing programmed cell death. Additionally, the interaction of DAXX with inflammatory cytokines, such as TNF-α, can amplify inflammatory cascades, underscoring its role in inflammation-driven processes (Pergaris et al., 2023). The apoptotic regulatory function of NRBP1 is closely linked to immune homeostasis and serves as a key factor in suppressing autoimmunity and maintaining peripheral T-cell homeostasis. Furthermore, as a core molecule in cellular stress responses, NRBP1 modulates ion transport and cell volume (Amnekar et al., 2025), a physiological basis that supports biological processes including immune cell migration, antigen presentation, and inflammatory activation, thereby regulating the adaptability of the inflammatory microenvironment and enabling precise control of immune responses. The findings of the present study further corroborate these previous discoveries at the genetic level and complete the logical chain.
According to the gene set analysis, 34 gene sets were significantly associated with SCI. Notably, the cellular response to biotic stimulus exhibited a significantly low p-value of 9.23 × 10−9, followed by positive regulation of RNA metabolic processes (p = 1.32 × 10−8). The cellular response to biotic stimuli plays a pivotal role in the inflammatory process. When an organism encounters stimuli such as trauma, infections, or immunological reactions, it triggers the initiation of inflammatory processes (Antar et al., 2023; Jochum and Fritz, 1989). The regulation of the cellular response to biotic stimuli involves the interplay between transcription factors, including NF-κB and IRFs, and chromatin organization within cis-regulatory regions of inflammatory genes (Wang et al., 2022). Gaining insights into the mechanisms underlying the initiation and amplification of the cellular response, as well as the negative feedback loops that modulate this process, is crucial for effectively managing inflammation. Inflammation has been linked to RNA metabolic processes, with emerging evidence highlighting the role of long noncoding RNAs in the regulation of inflammatory and immune responses (Rashidmayvan et al., 2023). These long noncoding RNAs exert their influence on metabolic inflammation by modulating various metabolic pathways (Shi et al., 2020). Elucidating the functions of these genes may contribute to early diagnosis of this disease and could lead to the identification of potential therapeutic targets for a range of inflammation-associated diseases (Chini and Mandal, 2022).
Both tissue expression analysis of 30 general tissue types and 53 specific tissue types revealed significant associations between the spleen and SCI. The spleen serves as a vital immune organ within the body and plays a well-established role in host defense against bacteria, viruses, and other pathogens. Additionally, it functions as a blood filter, contributing to overall host health (Abdulloyevich et al., 2023; Alexandre and Mueller, 2023). The involvement of the spleen in both innate and adaptive immune responses has been widely recognized, as it can regulate local and systemic immunity (Bronte and Pittet, 2013). Recent research provides compelling evidence of a direct communication pathway between the brain and spleen, whereby the spleen can modulate humoral immune defense through specific brain regions, including corticotropin-related neurons in the paraventricular nucleus and the central nucleus of the amygdala (Zhang et al., 2020). The splenic sympathetic anti-inflammatory pathway involves the activation of the splenic sympathetic nerve through a synergistic effect between the vagus nerve and the autonomic nervous system (Nash et al., 2022). At the distal end of the splenic nerve, norepinephrine binds to the β2 adrenergic receptor on splenic lymphocytes, leading to the release of acetylcholine (Ache). Subsequently, Ache inhibits the release of TNFα by spleen macrophages via the α-7-nicotinic Ache receptor (Zhou et al., 2022). Understanding these pathways holds significant therapeutic potential, as they can be targeted in various ways to stimulate anti-inflammatory regulation in TNFα-related inflammatory diseases (Bonaz et al., 2021; Pu et al., 2022).
According to the gene set analysis, hemopoiesis exhibited a statistically significant p-value of 5.54 × 10−8. Consistent with these findings, our gene analysis highlighted a distinct genetic association with the LEPR gene, which is involved in a novel hematopoietic pathway that is essential for normal lymphopoiesis. Notably, lymphocytes play a pivotal role in the anti-inflammatory response. Previous studies have confirmed that the abnormal activation of hematopoietic-derived innate immune cells, such as neutrophils, is a core mechanism underlying chronic low-grade inflammation, and their dysfunction can directly lead to elevated levels of inflammatory indices, including the NLR. The enrichment of hematopoietic-related pathways and splenic immune signals identified in our GWAS further supports the notion that the hematopoietic system contributes to SCIby regulating the differentiation and function of immune cells (van de Vyver, 2023).
A recent study by Jia et al. (2026) characterized the plasma proteomic signature of frailty in 50,506 adults, identified novel functional modules implicated in frailty pathogenesis, and recognized key causal proteins including MMP1 and LGALS8. This study indicated that inflammatory pathways were the most significantly enriched pathways for frailty, metabolic dysfunction represented a core early driving event of frailty, and oxidative stress exhibited cross-regulation with inflammatory and metabolic pathways. In contrast to this large-scale population-based proteomic study, the present study extends these findings by identifying critical genetic loci for SCI through genome-wide association analysis, revealing the regulatory roles of immune organs such as the spleen, and characterizing core inflammatory pathways, thereby providing complementary genetic evidence for the mechanistic link between SCI and frailty. Both studies consistently demonstrate that chronic inflammation is an important driver of frailty. In addition, the present study further reveals that the APOE, LEPR, and PDE4B genes may promote frailty by regulating inflammatory processes.
Our study has several key strengths, including the use of a large sample size from the UK Biobank, and this is the first instance in which a GWAS of INFLA-scores has been utilized for this purpose. The results of this study will serve as a valuable foundation for the subsequent critical stages of research on the genetics and underlying factors of SCI. These findings will help investigators conduct the power calculations necessary for future studies and offer potential loci for further examination in replication studies.
However, it is crucial to acknowledge the limitations of our study. First, the INFLA-score captures a specific snapshot of an individual’s chronic inflammation status at a particular moment and may not encompass all facets of inflammation. Second, potential selection bias may arise due to the middle-aged and older (37–73 years) demographic characteristics of the UK Biobank participants, potentially leading to an overestimation of the observed chronic inflammatory state in this study, and some potential confounders, such as acute infection, detailed medication use, and comprehensive lifestyle factors, were not fully available or could not be accurately assessed in the UK Biobank dataset, which may lead to residual confounding. Finally, the analyses were restricted to white British participants to minimize confounding from population stratification, which may limit the generalizability of the findings to other ethnic groups, and did not establish a replication cohort, which compromises the accuracy of the research findings. Besides, though the current article focuses on a foundational GWAS of the INFLA score, we fully recognize the importance of exploring causal relationships with related traits. Integrating analytical approaches such as Mendelian randomization or colocalization analysis would be valuable for elucidating these causal links. Considering the methodological complexity of these analyses and the requirement for rigorous validation, we plan to address this limitation in a follow-up study specifically dedicated to causal inference and pathway enrichment analysis.
Conclusion
In conclusion, our findings indicate genetic correlations between SCI and several inflammation and cellular regulation traits. Additionally, our results suggest a close relationship between SCI and spleen tissues. As the first GWAS to apply an INFLA-score to assess chronic inflammation, these findings will help investigators conduct the power calculations necessary for future studies and offer potential loci for further examination in replication studies.
Authors’ Contributions
J.X. and W.S. conceived and designed the research; J.X., W.S., and P.L. conducted the analyses and wrote the article; P.L., J.W., and Y.W. interpreted the results; J.W. and W.S. contributed to the analysis; J.X., W.S., P.L., J.W., and Y.W. critically revised the article. All authors have read and approved the final article.
Ethics Approval and Consent to Participate
UK Biobank received ethical approval from the National Health Service National Research Ethics Service (reference 11/NW/0382).
Availability of Data and Materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Footnotes
Acknowledgments
The authors would like to thank all participants of the UK Biobank cohort who have provided necessary genetic and phenotypic information.
Author Disclosure Statement
The authors declare no competing interests.
Funding Information
This work was supported by the