
Abstract
Coronavirus disease 2019 and pulmonary arterial hypertension (PAH) are clinically distinct disorders that converge on severe pulmonary vascular dysfunction, endothelial injury, and cardiopulmonary failure. However, the shared systems-level molecular architecture linking acute, virus-induced vascular damage with chronic pulmonary vascular remodeling remains undefined. To address this critical gap, we conducted a novel, integrative multilayered network analysis. By simultaneously combining transcriptome profiles of lung tissue samples with protein–protein interaction, metabolic, and regulatory networks, we systematically compared the previously unmapped molecular landscapes of both conditions. Despite their distinct upstream triggers—immune receptor–dominated signaling in acute severe acute respiratory syndrome coronavirus 2 infection versus remodeling- and ion channel-associated signaling in PAH—cross-disease integration revealed a highly structured, convergent immunometabolic regulatory architecture. This core is defined by extensive transcriptional reprogramming and the coordinated rewiring of oxidative phosphorylation and acetyl-CoA–associated metabolism. Taken together, these findings define a systems-level convergence model that mechanistically links acute viral endothelial injury and chronic pulmonary vasculopathy through a shared immunometabolic axis. This integrative molecular framework provides a new foundation for prioritizing candidate biomarkers and therapeutic nodes that target the overlapping vascular and inflammatory mechanisms of both conditions.
Keywords
Introduction
The coronavirus disease 2019 (COVID-19) pandemic, caused by infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has had a profound global impact, particularly on the respiratory and vascular systems (Synowiec et al., 2021). While the primary manifestation of COVID-19 is acute respiratory distress syndrome, emerging evidence highlights its systemic effects, including endothelial dysfunction, hypercoagulability, and pulmonary vascular complications (Vassiliou et al., 2020). These vascular abnormalities bear striking resemblance to the pathophysiology of pulmonary arterial hypertension (PAH), a chronic and progressive disease characterized by vascular remodeling, elevated pulmonary arterial pressure, and right heart failure (Naeije et al., 2022). PAH is driven by endothelial dysfunction, inflammation, and dysregulated cellular proliferation, leading to impaired blood flow and hypoxia (Hudson and Farkas, 2021; Santos-Gomes et al., 2022). Notably, COVID-19 has been associated with pulmonary vascular complications, such as thromboembolism and microvascular injury, raising concerns about its potential to exacerbate or trigger PAH in susceptible individuals (Riou et al., 2024; Rodríguez et al., 2021). Furthermore, PAH patients may face heightened risks of severe COVID-19 outcomes due to their compromised cardiopulmonary function (Farmakis and Giannakoulas, 2023; Ryan et al., 2020). This intersection between COVID-19 and PAH underscores the need to explore their shared pathophysiological mechanisms.
Clinically, COVID-19 and PAH exhibit overlapping features, including endothelial dysfunction, inflammation, and hypoxia (Tian and Cen, 2023; Wang and Loscalzo, 2023). In COVID-19, viral entry via the angiotensin-converting enzyme 2 receptor disrupts endothelial integrity, triggering a cascade of inflammatory and thrombotic events (Jin et al., 2020). Similarly, PAH is marked by endothelial injury, leading to the release of pro-inflammatory cytokines, growth factors, and vasoactive mediators that drive vascular remodeling (Berghausen et al., 2019; Guignabert et al., 2024). At the molecular level, both diseases involve dysregulated pathways related to inflammation, immune response, and oxidative stress; however, the precise molecular mechanisms underlying their connection remain poorly understood (Wang and Loscalzo, 2023). Despite overlapping vascular phenotypes, no study has systematically compared the multilayered molecular architecture of acute viral pulmonary injury and chronic pulmonary vascular disease.
Although extensive research has been conducted on the severity of both conditions, relatively few studies have explored their shared molecular mechanisms (Tian and Cen, 2023). Most studies focus on clinical outcomes or isolated biological pathways, leaving a critical gap in our understanding of the systemic, integrative molecular landscape connecting these diseases. Network-based, multiomics approaches have successfully identified disease-specific molecular signatures and potential drug targets in PAH, demonstrating the effectiveness of integrative systems biology in rare pulmonary diseases (Kasavi, 2023). However, the application of these approaches to simultaneously study both diseases remains limited.
To date, no integrative multilayered analysis has simultaneously characterized the shared molecular architecture underlying both an acute viral pulmonary condition and a chronic vascular disease. Most studies have focused on clinical outcomes or isolated biological pathways. Conventional reductionist methodologies are frequently insufficient to capture the full complexity of these conditions, resulting in a significant gap in our comprehension of the systemic, integrative molecular continuum that bridges these diseases.
To model these hidden mechanisms, we hypothesized that despite their entirely distinct upstream triggers, both diseases share a convergent, mathematically definable immunometabolic regulatory network module. Therefore, we developed and applied a multilayered network framework to simultaneously map shared and disease-specific molecular architecture. By integrating transcriptomic data with protein–protein interaction (PPI), metabolic, and transcriptional regulatory networks, we utilized network topology measures alongside reporter feature algorithms to identify the key dysregulated pathways, shared regulatory modules, hub proteins, reporter metabolites, and previously unrecognized regulatory elements, such as coordinated microRNA modulation and potential therapeutic targets. This topological approach provides a systems-level perspective on the molecular interplay between an acute viral infection and a chronic vascular disease. Ultimately, our findings establish a robust computational convergence model that quantitatively links immunometabolic rewiring with vascular pathology, offering a new theoretical roadmap for understanding complex, overlapping cardiopulmonary systems.
Materials and Methods
Transcriptomic data processing and statistical modeling
Gene expression data representing the perturbed states of PAH and COVID-19 were acquired from the Gene Expression Omnibus (GEO) database (Barrett and Edgar, 2006). Microarray data GSE15197 containing lung tissue samples from 18 patients with PAH and 13 healthy controls, and RNA-sequencing (RNA-Seq) data GSE208076 containing lung tissue samples from 7 patients with COVID-19 and 3 healthy controls (Hosseini et al., 2023; Rajkumar et al., 2010), were systematically processed to map the transcriptomic reprogramming of each condition.
For the microarray data, expression values were quantile-normalized, and differentially expressed genes (DEGs) were identified using the GEO2R tool (www.ncbi.nlm.nih.gov/geo/geo2r/), which internally applies Linear Models for Microarray Data with empirical Bayes moderation of standard errors (Smyth et al., 2003). For the RNA-Seq dataset, differential expression was modeled assuming a negative binomial distribution using DESeq2 in R/Bioconductor (Love, 2021). To control the false discovery rate (FDR) across multiple hypotheses, p values were adjusted using the Benjamini–Hochberg procedure. A stringent statistical threshold (adj. p value <0.05 and |fold change| >1.5) was applied to both datasets to isolate significant DEGs.
Functional overrepresentation within this filtered gene set was determined using ConsensusPath (Kamburov et al., 2013), mapping DEGs to Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation sources (Ashburner et al., 2000; Kanehisa and Goto, 2000). The p values were determined by Fisher’s exact test and controlled by FDR. A significance level of 0.05 (adj. p value <0.05) was used to determine significant GO biological process terms and KEGG pathways.
Topological reconstruction and centrality analysis of PPI networks
To identify structurally critical nodes mediating the disease states, global PPI networks were computationally reconstructed. Physical interaction data (122,040 interactions and 11,198 proteins) were extracted from the Biological General Repository for Interaction Datasets (BioGRID) database (release 4.4.225) (Stark et al., 2006). Redundant and duplicate interactions were removed prior to network construction to ensure each protein pair was represented by a single unique interaction. Disease-specific sub-networks were generated by mapping the translated DEG-encoded proteins and their first-degree interacting neighbors (defined as proteins with a direct physical interaction in the BioGRID network) onto this global topology. The structural importance of individual nodes was quantified using two primary network topology metrics: degree centrality (the number of direct edges connected to a node, k) and betweenness centrality [the fraction of all shortest paths passing through a given node, CB(v)], using CytoHubba plugin of Cytoscape (v.3.9.1) (Chin et al., 2014; Shannon et al., 2003). Hub proteins were formally defined as the top 10 nodes exhibiting the highest combined degree and betweenness centrality, indicating their role as critical bottlenecks for information flow within the network.
Genome-scale metabolic modeling and algorithmic identification of reporter metabolites
To map the downstream metabolic consequences of the observed transcriptional reprogramming, we integrated the transcriptomic data with Human1, a comprehensive consensus genome-scale metabolic model comprising a bipartite graph of 13,417 reactions, 10,138 metabolites, and 3625 genes (Robinson et al., 2020). We employed the Reporter Features algorithm (Oliveira et al., 2008; Patil and Nielsen, 2005) to identify subnetworks around which the most significant transcriptional changes occurred. This algorithm operates by mapping the gene-level transcriptional changes (p values) onto the bipartite network (i.e., metabolic network) via the stoichiometric matrix. It converts gene-level p values into Z-scores and aggregates them around adjacent nodes (i.e., metabolites) to identify subnetwork regions exhibiting significant, coordinated transcriptional shifts. The algorithm was implemented via COnstraint-Based Reconstruction and Analysis (COBRA) Toolbox in MATLAB, and the metabolic model was optimized using the Gurobi Solver within the COBRA Toolbox framework (Heirendt et al., 2019; Vlassis et al., 2014). Metabolites exhibiting an aggregated Z-score corresponding to a p value of <0.05 were classified as reporter metabolites. Subsequent pathway enrichment analyses of these metabolites were conducted using the Metabolites Biological Role (v2.0) (Lopez-Ibanez et al., 2016), and significantly associated KEGG pathways were determined based on an FDR-corrected p value threshold of 0.05.
Identification of reporter receptors, transcription factors, and microRNAs
To trace the upstream regulatory architecture governing the transcriptomic state, directed multipartite graphs of transcriptional regulation were constructed. Experimentally validated transcription factor (TF)–target and microRNA (miRNA)–target interactions were aggregated from Human Transcriptional Regulation Interactions, Transcriptional Regulatory Relationships Unraveled by Sentence-based Text mining, and miRTarBase (miRTarBase 9.0) databases (Bovolenta et al., 2012; Han et al., 2018; Huang et al., 2022). Similarly, a receptor–protein interaction network was built by extracting physical PPIs of proteins possessing GO-defined receptor activity (GO: 0004872) and its 304 child terms obtained from Database for Annotation, Visualization, and Integrated Discovery, Gene Ontology, Gene Set Enrichment Analysis, and Genecodis databases (Garcia-Moreno et al., 2022; Huntley et al., 2014; Mootha et al., 2003; Sherman et al., 2022; Subramanian et al., 2005).
The Reporter Features algorithm (Oliveira et al., 2008; Patil and Nielsen, 2005) was subsequently applied to these directed networks to calculate the cumulative statistical significance of the subgraphs surrounding each TF, miRNA, and receptor. Nodes meeting the p value <0.05 threshold were designated as reporter regulatory elements, representing the core upstream drivers of the observed disease states.
Results
Global transcriptomic reprogramming and cross-disease overlap
The gene expression datasets for PAH and COVID-19 were analyzed individually to identify DEGs and their expression patterns. Statistical analyses revealed numerous genes with significant changes in their expression levels (adj. p value <0.05 and |fold change| >1.5), including 103 commonly upregulated and 71 commonly downregulated DEGs between COVID-19 and PAH (Fig. 1A). A total of 1288 genes, 777 upregulated and 511 downregulated, were identified as DEGs in COVID-19 (Fig. 1B). In PAH, 2944 genes were upregulated, whereas 2453 genes were downregulated (Fig. 1C).
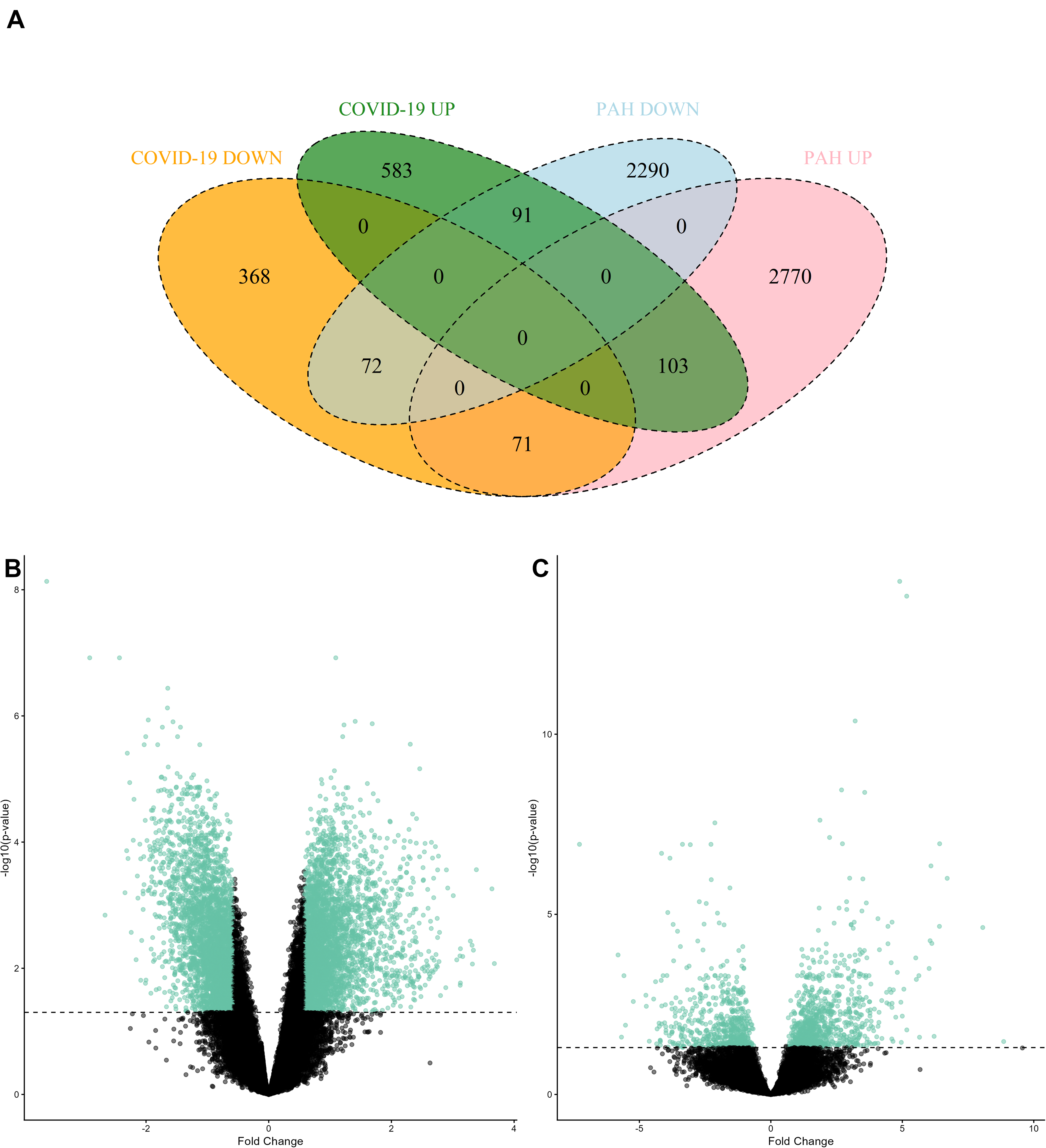
Overrepresentation analyses were performed to elucidate molecular pathways and biological processes associated with DEGs (Fig. 2). Pathway enrichment analysis indicated that neuroactive ligand–receptor interaction, Wnt signaling pathway, and basal cell carcinoma were upregulated, while herpes simplex virus 1 infection and messenger RNA surveillance pathway were downregulated in PAH. In COVID-19, the upregulated DEGs were significantly related to immune system-associated pathways, oxidative phosphorylation (OxPhos), cardiomyopathy, and several neurodegenerative diseases. On the contrary, the downregulated DEGs were significantly enriched for tumor necrosis factor (TNF) and interleukin (IL)-17 signaling pathways.
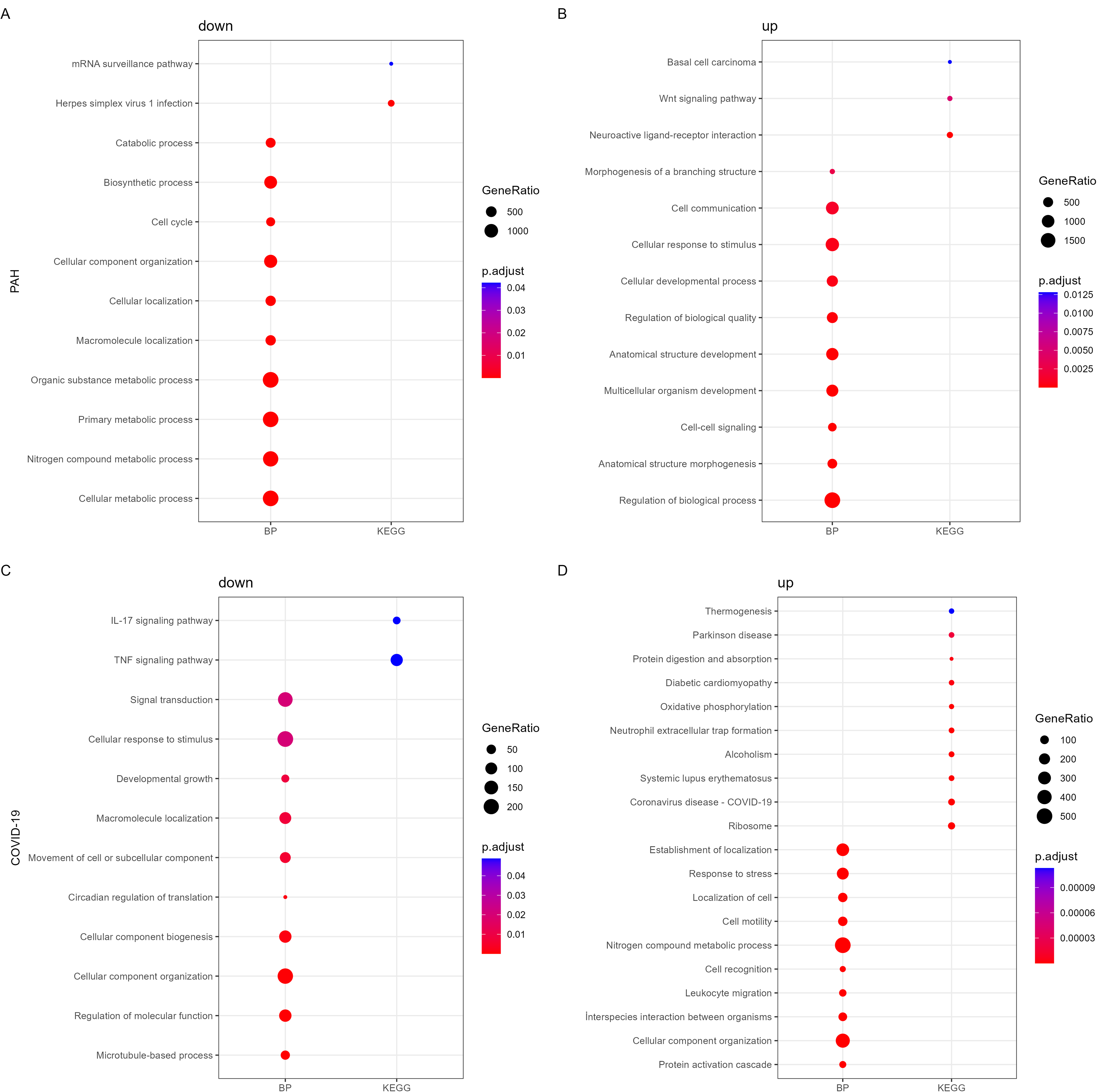
KEGG pathway and GO BP enrichment analyses of DEGs that were
The functional enrichment analysis of DEGs revealed the upregulation of cell–cell signaling and cell communication, and downregulation of cell cycle and cell localization in PAH. In the presence of COVID-19, immune response, leukocyte migration, production of molecular mediators of immune response, cell motility, cell activation, cellular homeostasis, and oxidation–reduction process were upregulated, whereas cell communication, cell growth, and respiratory gaseous exchange were downregulated. Rather than analyzing these pathways in isolation, these extensive transcriptomic shifts were utilized as the primary input parameters for our multilayered network integration. By quantifying these expression changes, we established the initial biological state required to map downstream topological perturbations within the human interactome and genome-scale metabolic networks.
Network topology reveals distinct hub proteins but shared regulatory architecture
To identify hub proteins, PPI networks around the proteins encoded by DEGs were reconstructed. The PPI network of COVID-19 consisted of 4901 proteins, including 800 proteins encoded by DEGs and their first neighbors, with a total of 18,440 physical interactions between these proteins. The PPI network of PAH comprised 8995 proteins, including 3325 proteins encoded by DEGs and their first neighbors, with a total of 61,969 physical interactions between these proteins. To identify critical information bottlenecks and highly connected regulatory coordinators, we analyzed the reconstructed PPI networks using graph theory metrics. The 10 proteins exhibiting the highest degree (indicating extensive local connectivity) and betweenness centrality (indicating crucial bridging roles between distinct subnetworks) were defined as structural hubs, and those that were encoded by DEGs were further analyzed (Supplementary Table S1). In PAH, four proteins (MYC, BRD4, YWHAG, and EP300) encoded by upregulated genes and six proteins (CUL3, KRAS, YWHAH, YWHAE, HSPA5, and LMNA) encoded by downregulated genes were identified as hubs. On the contrary, eight proteins (HSPA1A, CLPP, LAMP1, APP, RPS6, DYNLL1, RPL11, and RPL31) encoded by upregulated genes and two proteins (SRC and WWP2) encoded by downregulated genes were considered hubs in COVID-19.
Immunometabolic convergence module
By mapping the transcriptomic data onto the bipartite genome-scale metabolic network using the Reporter Features algorithm, we identified subnetwork regions exhibiting highly coordinated transcriptional shifts. This topological integration revealed that the most significant perturbations occurred around 108 and 115 reporter metabolites in the presence of COVID-19 and PAH, respectively. Reporter metabolites commonly identified in both diseases included PI pool, uridine diphosphate-N-acetyl glucosamine, D-galactosyl-N-acylsphingosine, and N-acetylneuraminate. These metabolites play roles in beta-oxidation of unsaturated fatty acids and N-glycan metabolism.
Functional enrichment analyses of reporter metabolites were performed to elucidate alterations and differences in the metabolic activities in response to COVID-19 and PAH (Fig. 3; Supplementary Table S2). Oxidative phosphorylation was commonly observed in both diseases. However, COVID-19 specifically affected aminoacyl-tRNA biosynthesis, Parkinson’s disease, endocytosis, and lysosome. Analysis highlighted several metabolic pathways (i.e., pyrimidine, purine, propanoate, sphingolipid, pyruvate, and glyoxylate and dicarboxylate metabolism); steroid biosynthesis; valine, leucine, and isoleucine degradation; synthesis and degradation of ketone bodies; tricarboxylic acid cycle cycle; taste transduction; and insulin signaling pathways, specifically in PAH.
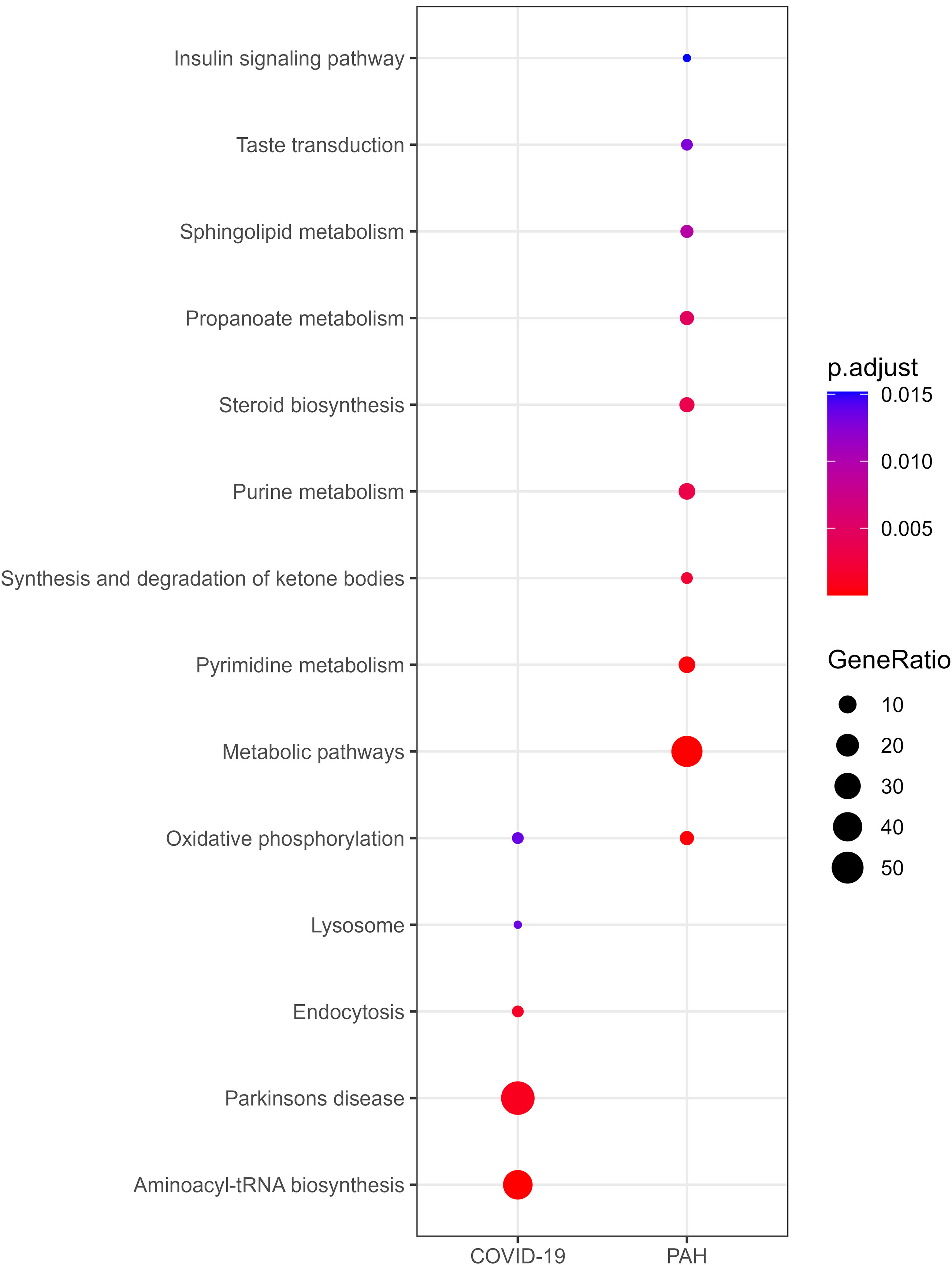
Overrepresentation analysis of reporter metabolites.
Manual inspection of metabolites that were not associated with a statistically significant pathway revealed distinct patterns of COVID-19 and PAH. In COVID-19, pathways involved in amino sugar and nucleotide sugar, sphingolipid, biopterin, folate, and sulfur metabolism, as well as pathways related to amino acid metabolism (i.e., lysine, cysteine, and methionine metabolism), glycan-associated pathways (i.e., N-glycan and O-glycan metabolism, and chondroitin/heparan sulfate biosynthesis), blood group biosynthesis, protein degradation, and keratan sulfate biosynthesis and degradation were identified. On the other hand, metabolites involved in lipid metabolism (i.e., fatty acid oxidation, bile acid and cholesterol biosynthesis), amino acid metabolism (i.e., arginine, proline, beta-alanine, tyrosine, valine, leucine, and isoleucine metabolism), and pathways associated with androgen, ascorbate and aldarate, glutathione, nicotinate and nicotinamide, nucleotide, purine, pyrimidine, pyruvate, steroid, vitamins C and D, and xenobiotics metabolism were observed in PAH. Metabolites that play roles in inositol phosphate and phosphatidylinositol phosphate metabolism were found in both diseases.
The reporter regulatory elements (i.e., TFs and miRNAs) were identified using the previously reconstructed human transcriptional regulatory network consisting of 25560 TF–gene interactions between 828 TFs and 12,678 genes, as well as 387836 miRNA–gene interactions between 2867 miRNAs and 15,424 genes (Akca and Kasavi, 2024). The analysis revealed that the most significant changes occurred around 6 TFs and 128 miRNAs in the presence of COVID-19. A total of 22 TFs and 358 miRNAs were identified as reporter regulatory elements that play roles in the transcriptional and post-transcriptional control of DEGs in PAH. No shared reporter TFs were found, but 20 miRNAs were commonly observed in both diseases. Our analysis indicated a shared immunological convergence of both diseases.
Divergent receptor signaling landscapes
To identify key signaling elements, the receptor–protein interaction network was reconstructed by extracting interactions of proteins with receptor activity. The reconstructed network consists of 10169 interactions between 4098 proteins, including 736 receptors and their interacting partners. A total of 23 and 28 proteins were identified as reporter receptors in the presence of COVID-19 and PAH, respectively.
The reporter receptors of COVID-19 contained proteins associated with the immune system (HLA-C, CLEC4D, CLEC4E, IL1R1, Toll-like receptor 4 [TLR4], LGALS3BP, KIR2DS2, C5AR1, TNFRSF10B, TNFRSF13B, TNFRSF13C, and TNFRSF17), as well as proteins that have roles in transport activity (CSE1L, IPO4, IPO5, IPO11, RAN, RANBP6, and XPO4). The reporter receptors of PAH included proteins associated with cancer and cell adhesive properties (AMFR, INTS6, KISS1R, XPR1, PTPRE, GP1BB, and EFEMP1), that play roles in the nervous system (CELSR3, GABBR2, GFRA1, GFRA2, GPR37, and EPHB3), involved in the immune system (IL9R, IL1R2, CLEC2D, and GPR35), and that function as ion channels (KCNJ3, PTK2B, and PKD2). Moreover, CRY2, which regulates the circadian clock; AGTR1, a vasoconstricting peptide that acts as a key regulator of blood pressure; and KCNJ3, which plays an important role in regulating heartbeat, emerged as prominent features in PAH. Collectively, our analysis revealed an immune receptor–dominant architecture of COVID-19 and G protein–coupled receptor/ion channel–dominant landscape of PAH.
Integrated multilayered convergence model of COVID-19 and PAH
To define a unified systems framework that moves beyond parallel, disease-specific analyses, we constructed an integrated multilayered interaction model that summarizes the principal molecular axes identified across transcriptomic, proteomic, metabolic, and regulatory layers (Fig. 4). Moving beyond static interaction maps, we mathematically conceptualized both conditions as distinct topological perturbations at the network periphery that funnel into a shared, highly connected central bottleneck (immunometabolic and vascular regulatory modules).
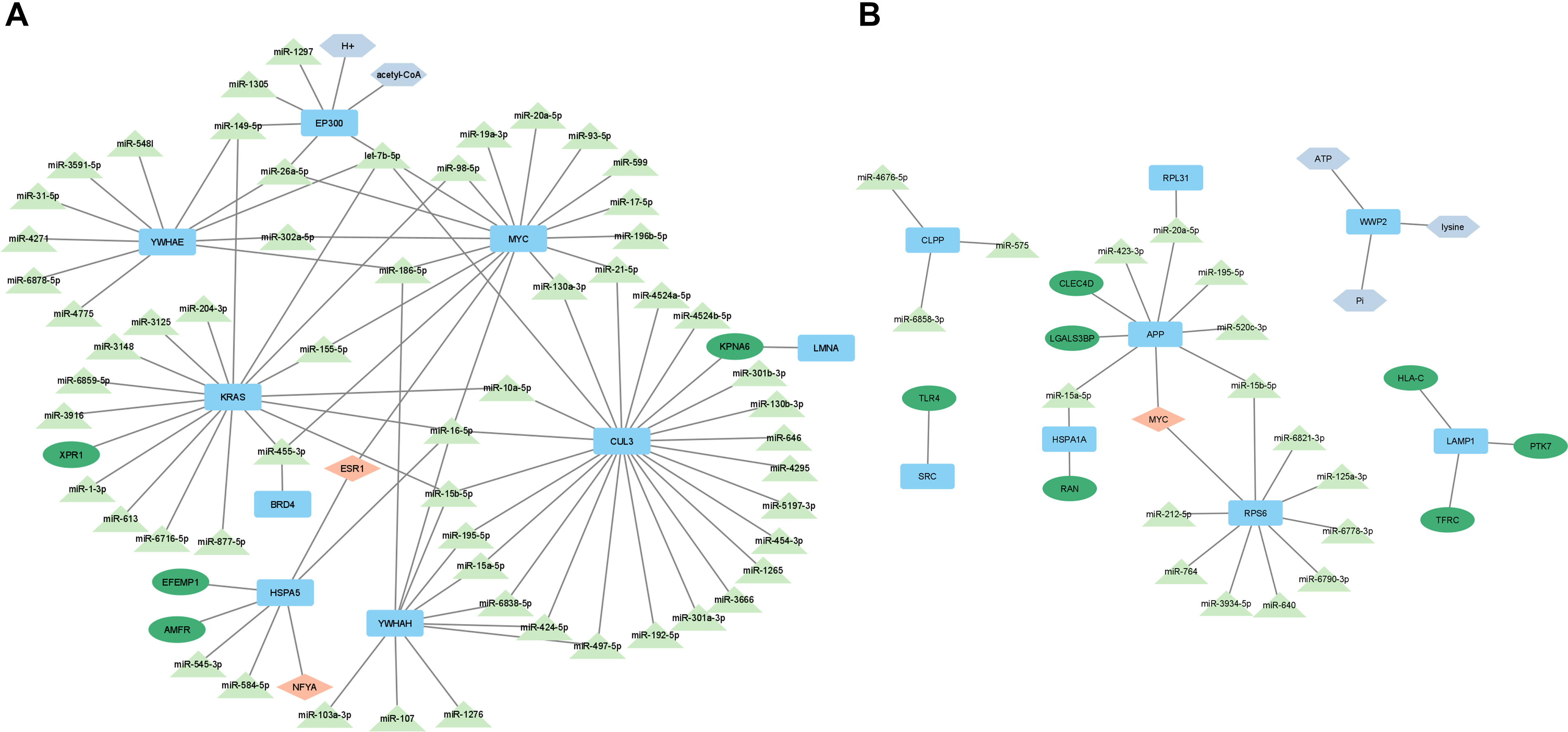
Comprehensive interaction maps of
At the upstream level, the characteristics of the immune response to SARS-CoV-2 were dominated by the activation of immune receptors, including pattern-recognition and antigen-presentation receptors. This reflects the acute inflammatory activation triggered by the virus. In contrast, PAH displayed prominent engagement of receptors and hubs associated with vascular remodeling, ion channel regulation, and proliferative signaling. These disease-specific entry nodes form distinct initiation layers within the network.
Despite their different triggers, both diseases converge on a central regulatory core defined by shared transcriptional and post-transcriptional modulators. The most notable of these are MYC-centered regulatory circuitry and overlapping miRNAs. This regulatory convergence is supported topologically by the positioning of high-centrality hubs within reconstructed PPI networks, indicating coordinated control over inflammatory, proliferative, and metabolic programs.
At the metabolic layer, reporter metabolite analysis revealed convergence on energy homeostasis and OxPhos pathways. Acetyl-CoA-associated reactions and ATP-related processes emerged as shared metabolic nodes. These results suggest a model of immunometabolic rewiring, in which inflammatory activation and endothelial stress are linked to changes in cellular energy utilization. Lysosomal and autophagy-related components further bridge immune activation and metabolic adaptation, reinforcing the presence of a coordinated stress response module.
Downstream of this shared regulatory-metabolic core, both conditions demonstrated enrichment of pathways governing endothelial dysfunction, cytokine amplification, vascular tone regulation, and oxidative stress. These convergent outputs establish a unified pathological axis that links acute endothelial injury caused by a viral infection in COVID-19 with chronic pulmonary vascular remodeling in PAH.
Discussion
While the clinical overlap between the cardiovascular complications of COVID-19 and PAH is increasingly recognized, the literature has lacked a unifying theoretical and computational model to explain this phenomenon. Current analytical strategies primarily rely on descriptive, isolated pathway analyses that fail to capture the multilayered complexity of the interactome. The primary conceptual advancement of this study is the development of an integrated, multilayered mechanistic framework based on network topology. We provide the first network-based mathematical mapping demonstrating that despite their fundamentally distinct etiologies—an acute viral infection versus chronic progressive vascular remodeling—both conditions funnel into a shared, highly structured immunometabolic convergence bottleneck (Fig. 5).
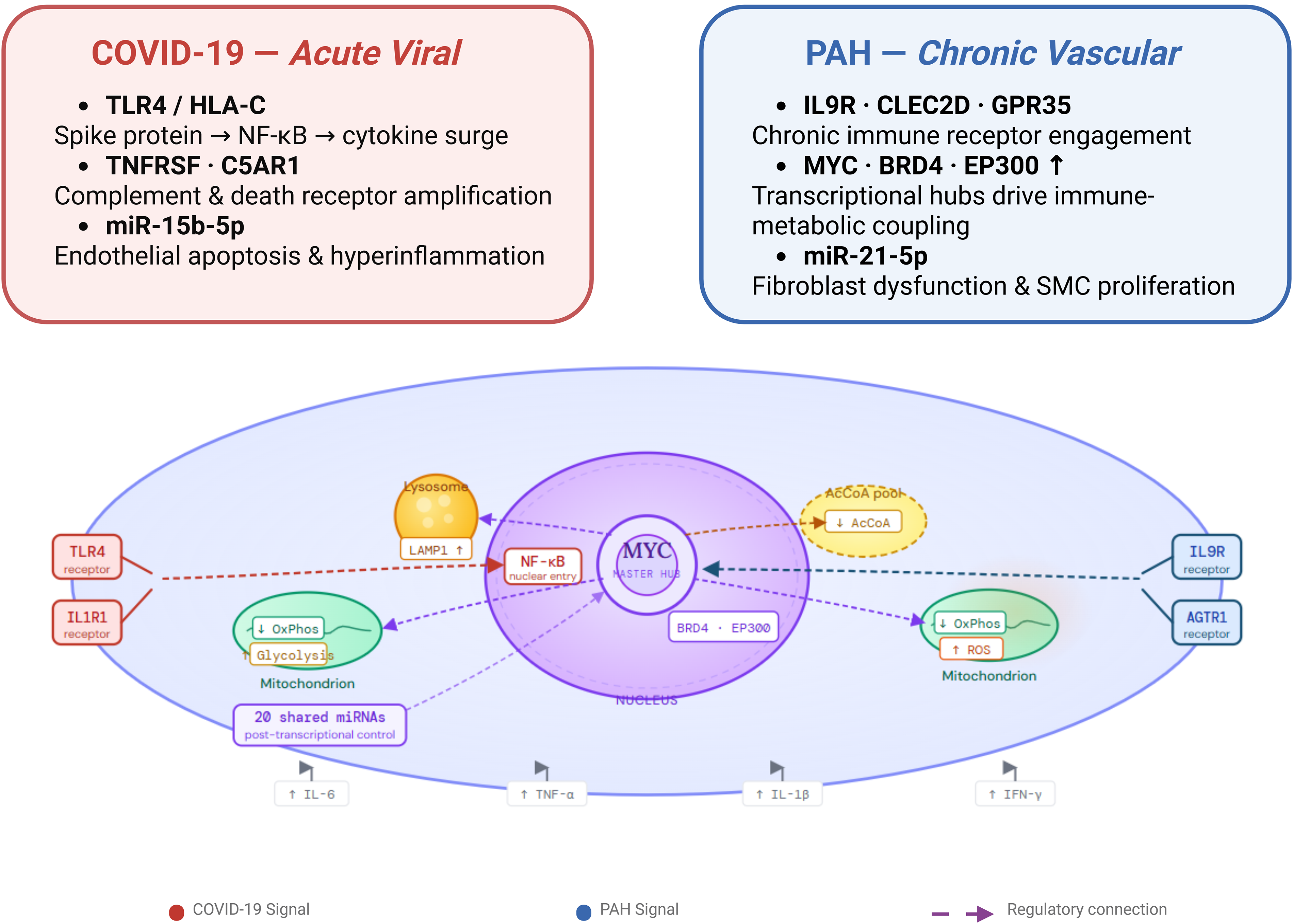
MYC-centered immunometabolic convergence between COVID-19 and PAH at the cellular level.
By integrating transcriptomic, proteomic, metabolic, and regulatory layers, we demonstrate that the observed overlap is not coincidental; rather, distinct upstream receptor-driven perturbations funnel into a central regulatory bottleneck. This systems-level integration mechanistically explains the overlapping endothelial dysfunction, inflammatory amplification, and cardiopulmonary impairment observed clinically, redefining how we view the relationship between acute viral endothelial injury and chronic pulmonary vasculopathy.
Cardiovascular convergence and vascular remodeling
Cardiovascular dysfunction emerged as a central convergent axis in our integrated network model, linking acute viral endothelial injury in COVID-19 with chronic vascular remodeling in PAH. Though the triggering factors differ, both conditions exhibited coordinated dysregulation across transcriptional, proteomic, and metabolic layers that collectively promote endothelial stress, vascular tone imbalance, and myocardial strain. Rather than representing independent cardiovascular complications, these findings suggest a shared systems-level vascular response characterized by inflammatory amplification coupled with metabolic disturbance.
At the regulatory level, several regulatory miRNAs and TFs converge on pathways that govern endothelial integrity and nitric oxide signaling. In COVID-19, regulatory miRNAs such as miR-15 family members and miR-195-5p are known to modulate angiogenesis and endothelial apoptosis. This may exacerbate microvascular injury. In PAH, miR-21-5p has been observed to promote fibroblast dysfunction and remodeling (Dingsdag et al., 2021; Giannella et al., 2022). Furthermore, regulators including TFs ESR1 and NFYA are implicated in endothelial homeostasis and vascular inflammation control (Clapauch et al., 2014; Desjardins and Balligand, 2006; Silvestre-Roig et al., 2013), suggesting compensatory or maladaptive transcriptional shifts within chronically stressed pulmonary arteries in PAH. Within the integrated interaction map, these regulatory elements have been positioned upstream of genes controlling vascular smooth muscle proliferation, extracellular matrix remodeling, and inflammatory mediator production, reinforcing their role in shaping vascular pathology.
At the protein level, disease-specific hubs revealed distinct entry points that converge on common cardiovascular outcomes. In COVID-19, central proteins such as HSPA1A and APP have been linked to oxidative stress responses and myocardial injury, which is consistent with reports of cardiac dysfunction associated with the virus (Cai et al., 2011; Chernyak et al., 2020; Knight et al., 2020; Li and Yang, 2021; Multhaup et al., 2002). In PAH, hubs including KPNA6 and AMFR have been associated with nuclear transport and endoplasmic reticulum stress. These processes have been implicated in vascular remodeling programs that drive right ventricular overload (Fu et al., 2011; Sun et al., 2011; Zhang et al., 2024). Despite differences in the identity of the upstream hubs, both networks feed into modules that govern oxidative stress, inflammatory cytokine production, and cellular survival signaling, underscoring convergence at the level of vascular stress-response pathways.
Metabolic integration further strengthened the shared cardiovascular axis. Reporter metabolite analysis revealed enrichment of energy-related intermediates, including acCoA and ATP-associated processes, highlighting altered bioenergetic demands in both diseases. These metabolites emerged due to their critical roles in energy balance, oxidative stress, and metabolic acidosis that worsen cardiovascular outcomes (Lopaschuk et al., 2021; Shu et al., 2023). Endothelial cells exposed to inflammatory or hypoxic stress undergo metabolic shifts that influence nitric oxide production, reactive oxygen species generation, and vascular tone regulation. The identification of OxPhos and energy homeostasis pathways in both conditions suggested that metabolic stress is an integral component, not merely a secondary consequence, of vascular dysfunction. In this context, immunometabolic rewiring directly interfaces with endothelial injury and vascular remodeling.
Together, these findings suggest that cardiovascular impairment is not merely an incidental overlap between COVID-19 and PAH, but rather a convergence outcome driven by coordinated regulatory, proteomic, and metabolic perturbations. By mapping these multilayered interactions, our study established a mechanistic link between acute inflammatory endothelial damage and chronic vascular remodeling within a unified cardiopulmonary systems model.
Immune dysregulation and immunometabolic rewiring
Immune dysregulation constituted a dominant convergence layer within the integrated network architecture, yet the nature of immune engagement differed at the level of upstream signaling while converging downstream on shared inflammatory and metabolic modules. In COVID-19, immune activation was predominantly receptor driven, characterized by enrichment of pattern-recognition and antigen-presentation receptors, consistent with viral-triggered innate and adaptive immune responses. In contrast, PAH exhibited a more chronic inflammatory profile, marked by regulatory shifts associated with vascular remodeling and sustained immune activation rather than acute pathogen sensing. Despite these distinct initiating contexts, both diseases converged on coordinated transcriptional programs that couple immune activation to metabolic stress and endothelial dysfunction.
A central feature of this convergence was the shared positioning of MYC as a common reporter TF in both diseases. MYC is a critical topological hub whose structural positioning within the bipartite network coordinates both transcriptomic and metabolic sub-modules. MYC is increasingly recognized as a master regulator of immune cell metabolic reprogramming, facilitating the glycolytic switch required for rapid proliferation and cytokine production (Liu et al., 2015; Zou et al., 2018). In COVID-19, such reprogramming may sustain antiviral responses but, when excessive, can potentiate cytokine amplification and tissue injury. In PAH, MYC-associated signaling has been linked to hyperproliferation of pulmonary vascular vessels (Yegambaram et al., 2025), suggesting that similar transcriptional circuits operate under chronic vascular stress. The identification of MYC in both conditions supports the concept of a common immunometabolic integrator bridging acute and chronic inflammation.
Post-transcriptional regulation further reinforced this convergence. Shared and disease-specific regulatory miRNAs modulate cytokine production, endothelial survival, and immune cell differentiation. In COVID-19, miR-15b-5p and miR-4676-5p drive hyperinflammation and immune cell dysfunction, while miR-204-3p in PAH suppresses cytokine production and smooth muscle proliferation, revealing a balance between inflammatory promotion and suppression across the diseases (Ghafouri-Fard et al., 2022; Koga et al., 2018; Martino et al., 2023; Zheng et al., 2021). Rather than acting in isolation, these miRNAs are embedded within densely connected regulatory modules that influence nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling, antigen presentation, and leukocyte migration. The coordinated dysregulation of these miRNA–target networks suggests that immune remodeling in both diseases is not stochastic but organized within structured regulatory circuits.
At the receptor level, divergence was more pronounced. The COVID-19 network was dominated by receptors such as TLR4 and HLA-associated components, reflecting direct engagement of viral sensing and antigen processing pathways. TLR4 recognizes viral proteins such as the SARS-CoV-2 spike and initiates downstream signaling through NF-κB and mitogen-activated protein kinase pathways, leading to severe cytokine release syndrome and acute respiratory distress (Kircheis and Planz, 2023; Shirato and Kizaki, 2021). HLA-C, a key modulator of T cell and natural killer cell activity, also influences susceptibility and severity of infection through its role in antigen presentation (Blais et al., 2011; Sips et al., 2016). In PAH, immune-related receptors coexisted with those linked to vascular tone regulation and cell adhesion, indicating integration of inflammatory signaling within a broader remodeling context. Nevertheless, both receptor landscapes ultimately fed into overlapping downstream effectors controlling cytokine release, oxidative stress responses, and endothelial activation.
Metabolic coupling provided an additional layer of immune convergence. Reporter metabolite analysis highlighted perturbations in energy metabolism and acetyl-CoA–related pathways, underscoring the tight interdependence between immune activation and bioenergetic adaptation. Activated immune cells undergo metabolic rewiring to sustain effector functions, and dysregulation of these pathways can amplify inflammatory injury (Erra DF et al., 2018; Hao et al., 2022; Riemann et al., 2016). The concurrent identification of OxPhos and energy homeostasis alterations in both diseases suggests that immune dysregulation is mechanistically linked to metabolic imbalance, reinforcing the integrated immunometabolic axis proposed in our convergence model.
A major novelty of our findings lies in the discovery that immune dysregulation in these two diseases is not merely a collection of overlapping inflammatory markers, but a highly coordinated regulatory circuit. While upstream immune engagement diverges in COVID-19 and PAH, our multilayered analysis revealed a previously unmapped downstream bottleneck. The shared positioning of MYC-centered circuitry and our identified panel of shared regulatory elements act as the master integrators bridging acute and chronic inflammation.
Neuroinflammatory and lysosomal stress signatures
Beyond vascular and immune convergence, our integrative analysis identified enrichment of molecular components that overlap with pathways frequently implicated in neurodegenerative disorders. Importantly, these findings suggested that COVID-19 or PAH are associated with activation of shared stress-response and lysosomal regulatory modules that are also characteristic of chronic neuroinflammation.
In COVID-19, hub proteins such as HSPA1A and LAMP1 were centrally positioned within the interaction network. HSPA1A regulates protein folding and aggregation (Dong et al., 2022; Fan et al., 2023), and LAMP1 regulates lysosomal function and autophagy—processes essential for maintaining cellular proteostasis under inflammatory and oxidative stress. Dysregulation of these pathways has been widely described in neurodegenerative conditions, but they are also fundamental components of systemic stress adaptation (Cheng et al., 2018; Sharoar et al., 2021). Similarly, the identification of lysosome- and autophagy-associated modules in our reporter analyses suggests enhanced cellular attempts to manage inflammatory and metabolic burden.
In PAH, regulatory elements such as ESR1 and extracellular matrix–associated proteins, such as EFEMP1, linked to cellular stress responses were enriched, reflecting chronic remodeling and sustained inflammatory activation, which have been reported to reduce neuronal damage in Alzheimer’s and Parkinson’s models (Chakrabarti et al., 2014; Duggan et al., 2024; Zhao et al., 2025). While certain molecules identified here have been studied in neurodegenerative contexts, their presence in our network more likely reflects shared mechanisms of oxidative stress, mitochondrial dysfunction, and chronic immune activation rather than disease-specific neuronal degeneration.
Taken together, these observations support the presence of a broader stress-response convergence layer characterized by proteostatic imbalance, lysosomal adaptation, and oxidative signaling. Such pathways may contribute to systemic inflammatory sequelae and potentially long-term complications, but further tissue-specific and longitudinal studies are required to clarify their clinical implications. Within our convergence framework, these signatures are best interpreted as manifestations of sustained immunometabolic stress rather than direct evidence of neurodegenerative pathology.
Translational and therapeutic implications
The identification of a shared immunometabolic–vascular convergence axis between COVID-19 and PAH has significant theoretical implications for network pharmacology and systems medicine. Rather than targeting isolated inflammatory mediators, our topological model suggested that computational perturbation of central network integrators may yield broader system-wide stability. In particular, the high-centrality positioning of MYC within both disease interactomes highlighted it as an optimal theoretical target and as a potential upstream leverage point capable of modulating downstream inflammatory and vascular dysfunction simultaneously.
Targeting immunometabolic coupling may represent a particularly promising strategy. The shared enrichment of OxPhos, acetyl-CoA–associated pathways, and energy homeostasis modules indicated that metabolic rewiring is tightly linked to inflammatory amplification and endothelial stress. Pharmacological interventions that modulate cellular metabolism, such as regulators of mitochondrial function, glycolytic flux, or acetyl-CoA utilization, could therefore attenuate both immune overactivation and vascular remodeling. This systems perspective supported the growing interest in metabolic therapies for inflammatory and vascular diseases.
At the receptor and signaling levels, disease-specific entry nodes offered opportunities for precision targeting. In COVID-19, immune receptor–driven pathways such as TLR-associated signaling may be prioritized to dampen acute hyperinflammation, whereas in PAH, interventions directed toward proliferative and remodeling-associated signaling cascades may be more appropriate. Importantly, the convergence downstream of these distinct entry points suggests that combination strategies targeting both upstream triggers and shared central modules could provide additive or synergistic benefits.
From a biomarker standpoint, the integrated regulatory and metabolic signatures identified here—including shared miRNA panels and central hub proteins—may serve as candidates for cross-disease risk stratification or monitoring of vascular inflammatory burden. While experimental validation is required, the systems-level prioritization provided by our network framework offers a rational starting point for selecting molecular targets with multilayered support.
The clinical and translational novelty of our convergence module is highly actionable. Rather than continuing the standard practice of targeting isolated, downstream inflammatory mediators, our network framework prioritizes central, cross-disease integrators. The high-centrality positioning of MYC and the shared metabolic rewiring (specifically OxPhos and acetyl-CoA utilization) provide a specific conceptual roadmap for rational drug repurposing. Interventions targeting this core immunometabolic coupling could theoretically attenuate both the acute hyperinflammation of COVID-19 and the chronic vascular remodeling of PAH simultaneously.
Conclusions
Our integrative multilayered network analysis demonstrates that COVID-19 and PAH, despite arising from distinct etiological origins, share a convergent immunometabolic–vascular regulatory architecture. Although their upstream triggers differ fundamentally—with viral receptor-mediated inflammation driving COVID-19 and chronic remodeling–associated signaling underlying PAH—both conditions funnel into common central modules defined by MYC-centered transcriptional control, metabolic reprogramming, and endothelial stress cascades. This molecular convergence establishes a mechanistic bridge between acute inflammatory injury and chronic pulmonary vascular dysfunction, while illuminating structured network vulnerabilities of potential diagnostic and therapeutic significance. It should be noted, however, that the present findings stem from computational integration of publicly accessible transcriptomic datasets and are therefore inherently hypothesis-generating in nature. Additionally, the COVID-19 RNA-seq dataset used in this study has a limited sample size (n = 7 COVID-19 patients, n = 3 controls), which may limit the generizability of the transcriptomic findings. It should also be noted that network-based analyses are inherently limited by the completeness of interaction databases, which may favor well-studied proteins and underrepresent less-characterized molecular players. Although cross-layer validation enhanced the robustness and confidence of our findings, experimental investigations and prospective clinical studies remain essential to establish causality and confirm translational relevance. Notwithstanding these limitations, our systems-level framework provides a conceptual groundwork for mechanistic exploration and the rational prioritization of therapeutic targets across the spectrum of acute viral and chronic pulmonary vascular diseases.
Authors’ Contributions
C.K. conceived the study. D.C.K. carried out the analysis. D.C.K. and C.K. wrote the article. All authors read, revised, and approved the final version of the article.
Footnotes
Acknowledgments
The fellowship awarded to Defne Cig (program no. 2210-C) by the Scientific and Technological Research Council of Türkiye (TUBITAK) is gratefully acknowledged.
Data Availability Statement
The gene expression datasets analyzed during the current study are publicly available from the NCBI Gene Expression Omnibus (GEO) repository under accession numbers GSE15197 and GSE208076. The reconstructed human metabolic and regulatory network models are derived from previously published and publicly accessible databases, as detailed in the
Ethical Approval
This article employed a computational analysis of publicly available gene expression data. Ethical committee approval was not required for the study.
Declaration of Generative Artificial Intelligence and Artificial Intelligence–Assisted Technologies in the Article Preparation Process
During the preparation of this work, the authors used Gemini in order to synthesize complex literature and DeepL in order to improve language and readability, and check for grammar and spelling. After using these tools/services, the authors reviewed and edited the content as needed and take full responsibility for the content of the published article.
Author Disclosure Statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.
Funding Information
No funding was received for this article.
Supplemental Material
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.