
Abstract
Understanding epistatic interactions, where mutations collectively influence viral fitness, is critical for predicting pathogen evolution. We present a hidden Markov model (HMM) framework that captures the temporal dynamics of epistatic relationships in SARS-CoV-2, addressing limitations of static network-based approaches. Our method models single amino acid variant pairs as a two-state system (linked/unlinked), with emission probabilities derived from linkage disequilibrium theory and transition probabilities optimized via the Baum–Welch algorithm. We implement permutation-based validation with temporal order noise reduction (>80% agreement across five iterations) to distinguish biological signals from stochastic noise. Applied to 2,192,008 spike protein sequences from the United States (March 2020–December 2021), our approach identified three classes of epistatic dynamics: permanent (0.3%), transient (0.3%), and oscillating (0.7%) linkages. Analysis of Alpha variant positions revealed 78% epistatic linkage compared with 1.3% across all spike protein position pairs, with 60% exhibiting oscillating patterns suggestive of frequency-dependent selection. We detected all 17 previously reported epistatic pairs plus 18 novel interactions, including critical connections between positions 69–70 and other functional sites. Notably, Alpha variant epistatic networks were detectable as early as April 2020, months before widespread circulation. Our framework scales to variant-wide analysis, revealing distinct patterns across variants: Delta (96% linkage, 71% oscillating) and Omicron (87% linkage, 56% oscillating). The computational pipeline, implemented with parallelized HMM training and Viterbi decoding, processes hundreds of thousands of position pairs efficiently. By transforming epistasis detection from static to temporal analysis, this work provides computational tools for early variant detection and demonstrates how probabilistic modeling can capture evolutionary dynamics in real-time genomic surveillance systems.
INTRODUCTION
Predicting viral evolution represents one of the most consequential challenges in contemporary life sciences, requiring a nuanced understanding of both stochastic and deterministic factors that drive genomic change (Lässig et al., 2017; Mohebbi et al., 2024). This predictive capability becomes particularly critical for emerging pathogens, where anticipating evolutionary trajectories directly impacts our ability to design effective countermeasures and control strategies (Icer Baykal et al., 2021).
Central to this challenge is the phenomenon of epistasis—interactions where the fitness effect of a mutation at one genomic site depends on the allelic state at another site (Gros et al., 2009). Epistasis is formally defined as the difference between the observed fitness
Epistatic interactions manifest as either positive or negative effects on viral evolution (Lyons and Lauring, 2018). Positive epistasis can facilitate the accumulation of beneficial mutations, enhancing viral adaptability, while negative epistasis restricts certain mutational combinations, limiting potential evolutionary pathways (Starr and Thornton, 2016). Understanding these dynamics is particularly relevant for rapidly evolving viruses like SARS-CoV-2, where epistatic relationships determine which variants emerge and persist (Mohebbi et al., 2023).
The ability to detect and characterize epistatic interactions in real time has profound implications for public health response. First, it enables predictive surveillance that can anticipate the emergence of new variants before they achieve significant prevalence, providing crucial lead time for vaccine updates and therapeutic adjustments (Domingo et al., 2019; Palmer et al., 2015; Weinreich et al.). Second, epistasis represents a dynamic force that shapes viral adaptation across multiple timescales—from rapid within-host evolution to longer population-level changes—requiring analytical approaches that can capture these temporal dimensions. Third, understanding the historical accumulation of coordinated mutations reveals the structural and functional constraints that have shaped SARS-CoV-2 evolution, informing more targeted intervention strategies.
Traditional prefix-based approach to modeling epistatic interactions have followed combinatorial population modeling of random point mutations (Mohebbi et al., 2024). Although this approach has provided foundational insight, it suffers from critical limitations: it is inherently static, requires complete datasets for analysis, and cannot capture the dynamic sequential dependencies characteristic of viral evolution. Consequently, it fails to detect emerging trends and struggles to predict the effects of novel mutations, limitations that significantly hamper its utility for real-time surveillance and early variant detection.
HMMs offer a compelling alternative approach to overcome these limitations. HMMs excel at capturing temporal and sequential dependencies that define how mutations arise and interact over time (Li, 2024). By focusing on local dependencies and recent data rather than averaging across entire datasets, HMMs can identify emerging evolutionary patterns and adapt to rapidly changing genetic landscapes (Yoon, 2009). Their ability to model conditional relationships between mutations enables a more nuanced understanding of evolutionary pathways—for example, how stabilizing SAVs could enable subsequent immune escape mutations. Furthermore, HMMs provide computational efficiency and scalability essential for real-time analysis of large genomic datasets, making them particularly suitable for anticipating the emergence of new viral variants.
In this study, we present an HMM-based approach for tracking the temporal evolution of linkage patterns between pairs of SAVs, which may indicate functional relationships through mutation co-occurrence. We aggregated data across multiple sampling periods to address inherent uncertainty in sampling times and to mitigate noise from variable sequencing frequencies. Unlike static measures, this dynamic approach enables precise detection of when epistatic interactions form or dissolve, providing unprecedented resolution of viral adaptation pathways.
Our approach not only documents the spatiotemporal distribution of epistatic interactions but also elucidates how these interactions evolve in response to selection pressures. The integration of HMMs represents a significant advancement over traditional methods by capturing dynamic evolutionary changes rather than static snapshots. The resulting insights have immediate applications for public health response—identifying SAV combinations that may compromise vaccine efficacy or signaling evolutionary shifts indicative of adaptation to population immunity. These capabilities are essential for guiding timely updates to vaccines and therapeutics, ultimately contributing to more effective management of current and future pandemic threats.
METHODS
Problem formulation
The fundamental problem we address is detecting and characterizing epistatic interactions between SAVs in SARS-CoV-2 genomes using time-series data. Given a dataset of viral sequences collected over time, we aim to identify pairs of genomic positions that exhibit nonrandom co-occurrence patterns that are indicative of functional relationships.
Formally, let
The core problem is determining whether the observed co-occurrence patterns between positions u and v arise from functional epistatic relations (linked state) or independent evolutionary processes (unlinked state). We frame this as inferring a sequence
Our approach employs a two-state hidden Markov model (HMM) to infer these hidden states, coupled with permutation testing for statistical validation and classification of temporal dynamics to reveal evolutionary patterns. Through this approach, we can identify not only which positions interact epistatically, but also when these interactions emerge, dissolve, or fluctuate in response to changing selective pressures.
HMM approach
We implemented a two-state HMM to detect and characterize epistatic interactions between pairs of genomic positions, shown in Figure 1. The model represents the relationship between two positions as being in either a “linked” or “unlinked” state, with transitions between states corresponding to evolutionary changes in epistatic relationships. Formally, our HMM is defined as
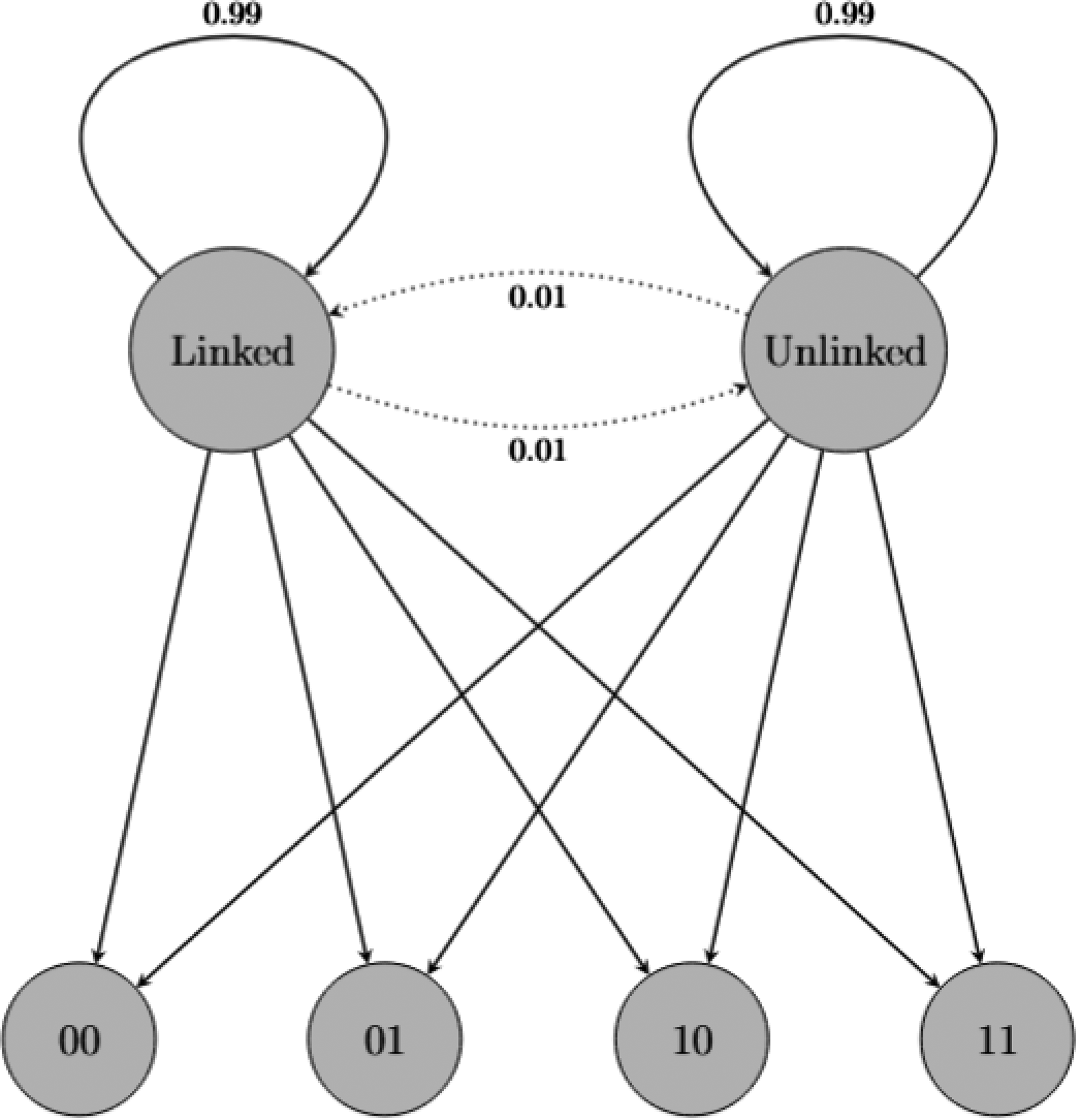
Two-state hidden Markov model for detecting epistatic interactions. The model consists of two hidden states: “Linked” (indicating epistatic relationship) and “Unlinked” (indicating independent evolution). The model emits observations {00, 01, 10, 11} representing the joint mutation state of two positions. Transition probabilities between states (shown on the connecting arrows) are initialized with a bias toward state persistence (0.99 for Linked→Linked, 0.99 for Unlinked→Unlinked) and are subsequently refined during training. Emission probabilities (paths from hidden states to observations) are fixed based on theoretical linkage disequilibrium calculations. The model’s parameter configuration is described in detail in the following section.
A represents the state transition probability matrix
B represents the emission probability matrix
The state space consists of two hidden states
The model is parameterized as follows:

Detailed parameter configurations for the transition and emission probabilities are described in the following sections, along with our approach to state sequence decoding and statistical validation.
For the initial state probabilities
The transition matrix A was initialized with a deliberate bias toward state persistence (0.99 probability of remaining in the linked state and 0.99 probability of remaining in the unlinked state). Unlike the emission matrix, these transition probabilities are not fixed but were further refined during training using the Baum–Welch algorithm (IEEE Information Theory Society, 2003) with five iterations and a convergence tolerance of 1e-5. The initial bias toward state persistence ensures the model learns to transition between states only when there is compelling evidence in the data, allowing us to identify meaningful evolutionary changes while minimizing spurious transitions due to sampling noise.
Emission probability calculation
The emission probabilities for each state were derived from the observed mutation patterns while accounting for linkage disequilibrium, a concept introduced by population geneticists to measure nonrandom associations between alleles at different genomic positions (Maynard and Haigh, 2007; Slatkin, 2008). For a pair of SAVs at genomic positions u and v, we calculate:
Individual mutation frequencies: Linkage disequilibrium coefficient D: This measures the deviation from the expected counts of co-occurrence if mutations were evolving independently (Lewontin, 1988). To establish this parameter, we first determine
For the “unlinked” state, emission probabilities we follow the independence assumption:
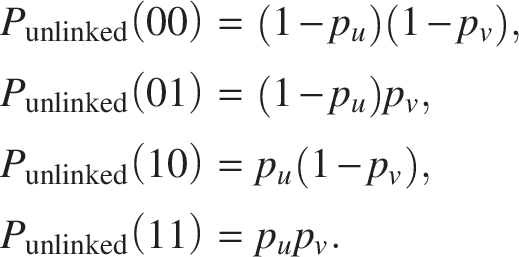
For the “linked” state, emission probabilities we incorporate the linkage disequilibrium parameter D:
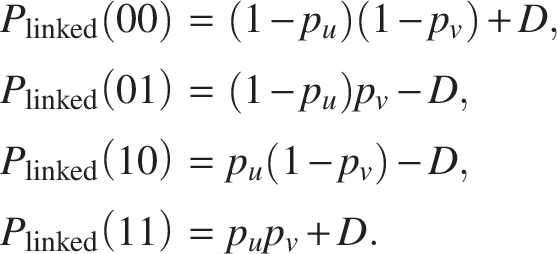
We determined the linkage disequilibrium parameter D by:

In our analysis, we set the value of
We implemented a customized HMM with fixed emission probabilities and trainable transition probabilities. The emission probabilities were calculated using the theoretical independence model described above (Lewontin, 1988) and remained fixed throughout the training process, while the transition probabilities were refined using the Baum–Welch algorithm (IEEE Information Theory Society, 2003).
For each SAV pair, we followed these steps:
Calculate and fix emission probabilities based on the observed frequency distributions and linkage disequilibrium parameters Initialize transition probabilities with a deliberate bias toward state persistence (0.99 for linked→linked, 0.99 for unlinked→unlinked) Apply the Baum-Welch algorithm to refine only the transition probabilities, using:
Five iterations maximum Convergence tolerance of 1e-5 Parameters configured to update only transition probabilities (“params = t”)
This approach allowed us to leverage prior knowledge about theoretical emission patterns while optimizing the transition dynamics based on the empirical data. By fixing the emission probabilities, we maintained consistency across all SAV pairs while still allowing the model to learn the appropriate transition behavior for each specific pair.
To analyze the temporal pattern of linkage for each SAV pair, we employed the Viterbi algorithm to determine the most likely sequence of hidden states given the observed mutation data. This provided a time series of linked/unlinked classifications that revealed when epistatic relationships emerged, dissolved, or persisted throughout the study period.
For each SAV pair
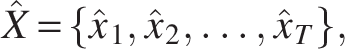
Statistical validation approach
To ensure the robustness of our findings and distinguish biologically meaningful epistatic interactions from random associations, we implemented a comprehensive statistical validation approach consisting of three key components:
Permutation testing
To generate a reliable null distribution, we performed controlled permutation testing that preserved the temporal structure of the data while disrupting potential linkages. Our permutation procedure employed two distinct temporal grouping strategies:
For each grouping strategy, we performed 5 independent permutations, creating a total of 10 randomized datasets. This approach preserved the overall mutation frequency dynamics within temporal windows while disrupting potential linkages between SAVs. Each permuted dataset underwent the same HMM analysis as the original data, resulting in five alternative state sequences for each SAV pair per grouping method.
To control for false positives and ensure reproducible detection of epistatic relationships, we implemented an order noise reduction approach that required consistent identification across multiple permutations:
For each SAV pair and temporal window, we counted the occurrences of each state (linked/unlinked) across all permutations We calculated the proportions of permutations supporting each state We applied a threshold (requiring
This approach effectively filtered out spurious associations that might appear by chance in individual permutations, ensuring that only robust and reproducible linkage patterns were identified.
Based on the temporal patterns of the most likely state sequences, we classified each SAV pair into one of three categories of evolutionary relevance:
This classification system provides a framework for interpreting the evolutionary significance of different epistatic interaction patterns and their potential contributions to viral adaptation.
RESULTS
Data
SARS-CoV-2 genomic sequence data were obtained from GISAID (Shu and McCauley, 2017). We focused on analyzing amino acid variants of the spike protein collected in the United States between March 2020 and December 2021. We utilized the method outlined in “Early detection of emerging viral variants through analysis of community structure of coordinated substitution networks” (Mohebbi et al., 2024) to create our dataset. Spike protein mutations were selected due to their critical role in identifying variants of concern (VOC) and their direct impact on viral fitness, transmissibility, and immune evasion properties (Davies et al., 2021). The final processed dataset contained
Baseline epistatic patterns across all spike protein positions
We first analyzed epistatic patterns across all possible pairs of spike protein positions to establish a baseline for comparison with variant-specific patterns. Among the 809,628 possible pairs formed by 1273 spike protein positions, only 10,850 pairs (1.3%) exhibited statistically significant linkage (Table 1). The distribution of these linked pairs revealed distinct temporal patterns: 2416 pairs (0.3%) showed permanent linkage, 2647 pairs (0.3%) demonstrated transient patterns, and 5787 pairs (0.7%) displayed oscillating dynamics. The vast majority, 798,778 pairs (98.7%), remained unlinked throughout the study period.
Distribution of Epistatic Relationship Patterns across Analyzed SAV Pairs in the SARS-CoV-2 Spike Protein Dataset (N = 2,192,008 Sequences)
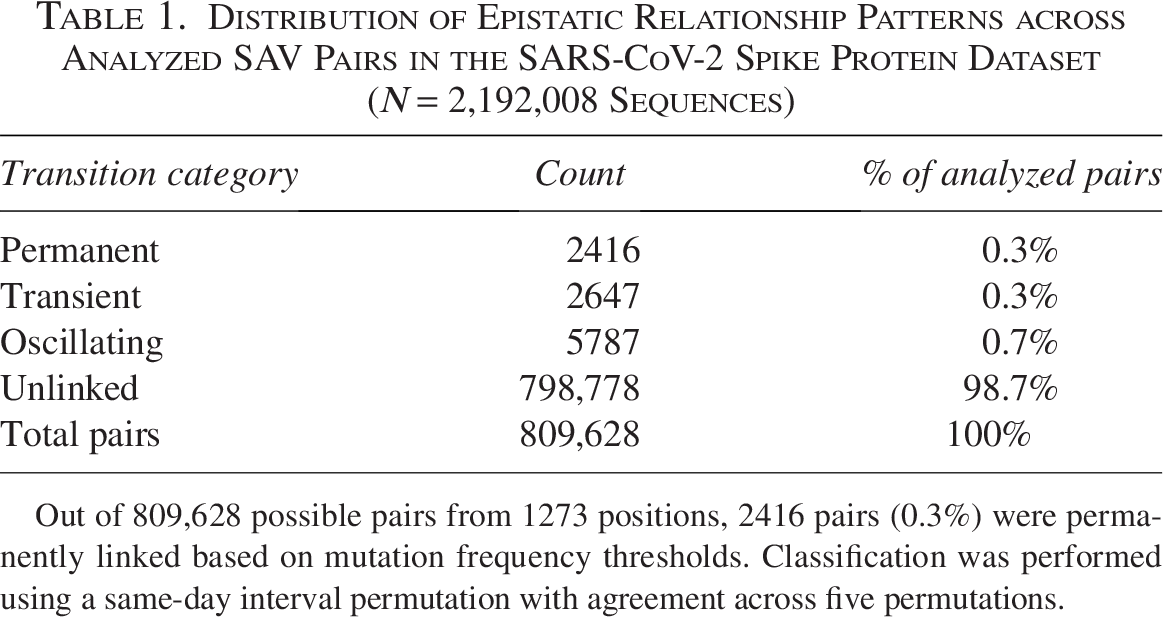
Distribution of Epistatic Relationship Patterns across Analyzed SAV Pairs in the SARS-CoV-2 Spike Protein Dataset (N = 2,192,008 Sequences)
Out of 809,628 possible pairs from 1273 positions, 2416 pairs (0.3%) were permanently linked based on mutation frequency thresholds. Classification was performed using a same-day interval permutation with agreement across five permutations.
This baseline distribution analysis reveals the rarity of stable epistatic relationships. The predominance of unlinked pairs underscores that epistatic interactions are highly selective and likely maintained only when conferring significant fitness advantages. These baseline frequencies provide crucial context for interpreting variant-specific patterns.
We applied our HMM approach to detect epistatic interactions across three major SARS-CoV-2 VOC: Alpha, Delta, and Omicron. The analysis was conducted in two phases. First, we performed an initial analysis without permutation to establish baseline linkage patterns. Subsequently, we implemented the permutation strategy described in the methods to generate robust null model distributions. For this validation, we employed two distinct temporal grouping methods: grouping sequences collected on the same day and grouping sequences within 14-day intervals, with five independent permutations for each method. We then applied temporal order noise reduction across these permutations to identify statistically significant epistatic relationships.
For the purposes of our analysis, we define a “statistically significant epistatic relationship” as a linked state between two SAVs that: (1) demonstrates strong evidence against the null hypothesis of independent evolution, (2) maintains its linked classification across at least 80% of permuted datasets, and (3) exhibits transition patterns that differ significantly from random fluctuations, as determined by our classification system for linkage dynamics.
A central hypothesis in viral evolution research is that SAVs of VOC are not merely co-occurring by chance but exhibit functional epistatic relationships that collectively enhance viral fitness through coordinated effects (Lyons and Lauring, 2018; Mohebbi et al., 2024). To test this hypothesis, we analyzed known mutation positions characteristic of each variant (Choi and Smith, 2021).
For the Alpha variant, among 45 possible pairs formed by 10 characteristic mutations, we identified 35 significantly linked pairs (78%). These included all 17 pairs previously identified by prefix-based approaches (Mohebbi et al., 2024) plus 18 additional pairs. The distribution revealed 3 pairs (7%) with permanent linkage, 5 pairs (11%) showing transient patterns, and 27 pairs (60%) exhibiting oscillating dynamics, while 10 pairs (22%) remained unlinked.
The Delta variant analysis examined 28 possible pairs from 8 key mutations, detecting 27 linked pairs (96%). Of these, 4 pairs (14%) exhibited permanent linkage, 3 pairs (11%) showed transient linkage, and 20 pairs (71%) displayed oscillating patterns, with only one pair (4%) remaining unlinked.
The Omicron variant presented substantially larger epistatic activity. Among 666 possible pairs from 37 key mutations, we identified 578 linked pairs (87%). The distribution differed notably from Alpha and Delta: 152 pairs (23%) showed permanent linkage, 50 pairs (8%) exhibited transient linkage, and 376 pairs (56%) demonstrated oscillating dynamics, while 88 pairs (13%) remained unlinked.
Figure 2 shows the detailed temporal dynamics of selected position pairs, revealing the distinct patterns of epistatic evolution across variants. Table 2?? provides the complete distribution of linkage categories.
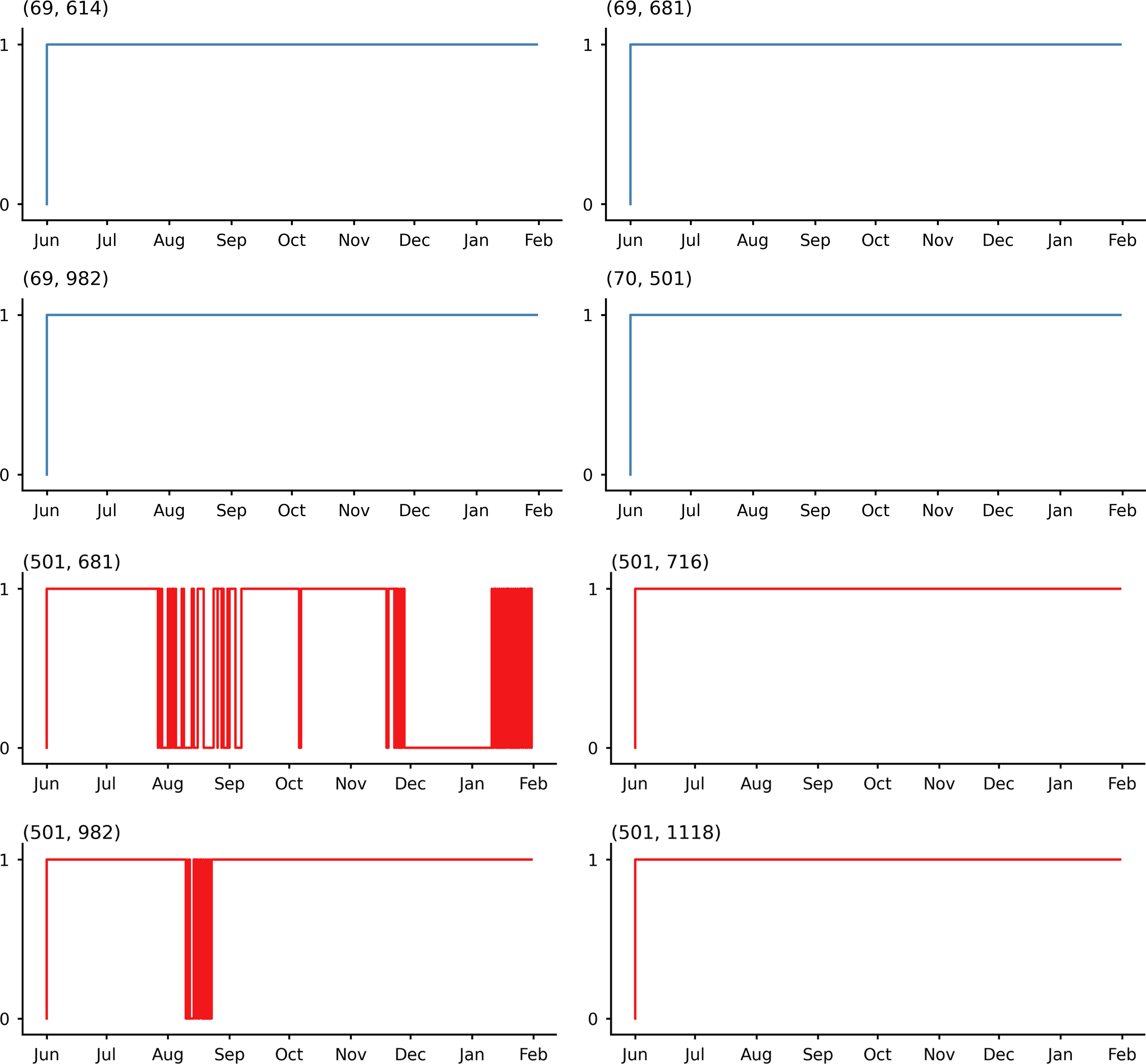
Temporal evolution of linkage states for Alpha variant position pairs. Each subfigure shows one pair’s transition from unlinked (0) to linked (1), over the same time axis from June 2020 to January 2021. These patterns were validated through temporal order noise reduction across 10 independent permutations, requiring >80% agreement for classification.
The Alpha variant (Fig. 3) showed the initial establishment of approximately 35 linked pairs in early 2020. This was followed by a gradual decline beginning in July 2020, with fluctuations between 20 and 30 linked pairs continuing through early 2021.
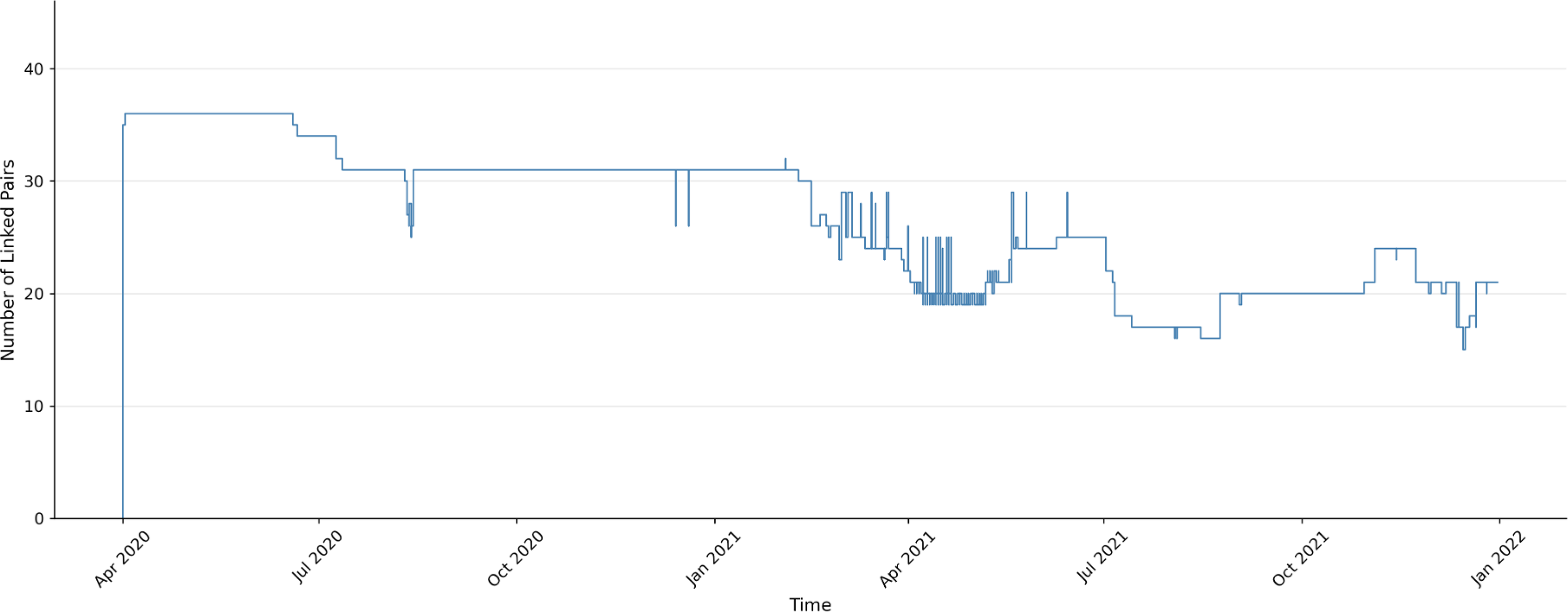
Linked pair dynamics for the Alpha variant from early 2020 through late 2021.
Delta variant linkages (Fig. 4) emerged later, with initial detection around April 2020. At that time, approximately 20 linked pairs were observed, which then decreased to around 15 pairs. This was followed by variable patterns ranging from 8 to 17 linked pairs through the remainder of 2021.
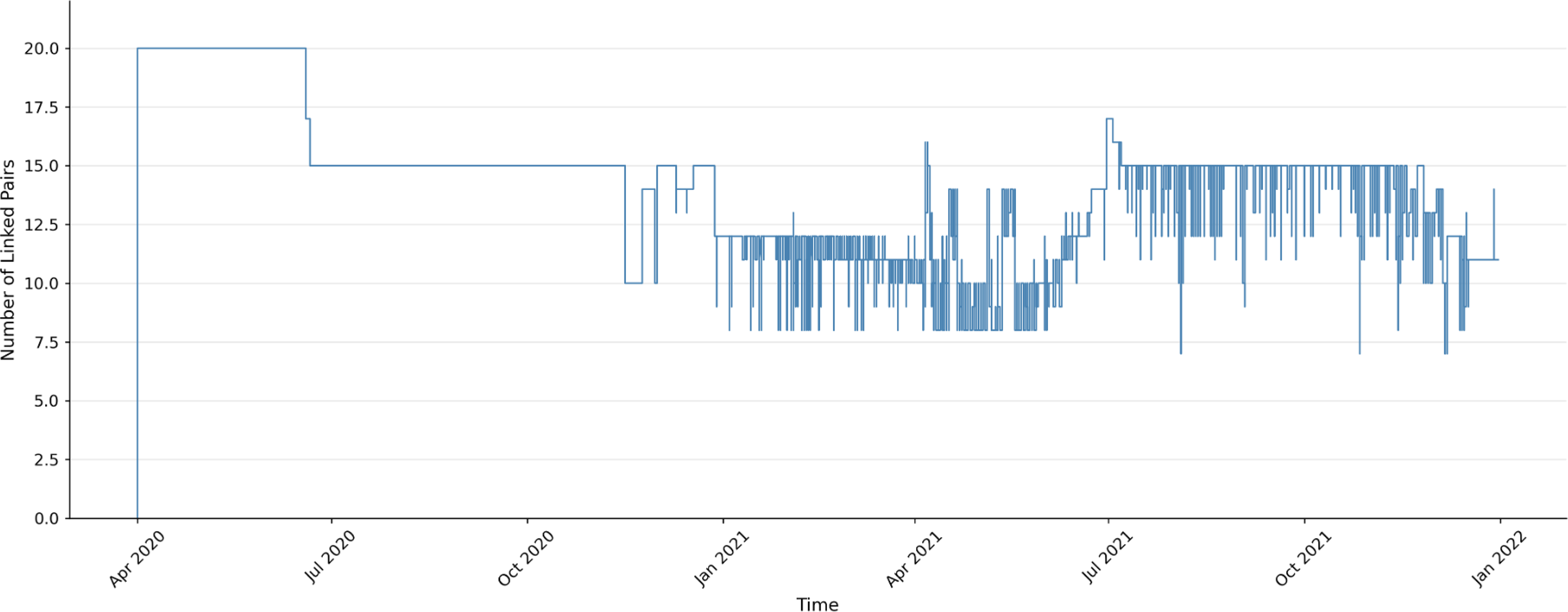
Linked pair dynamics for the Delta variant from April 2020 through 2021.
The Omicron variant (Fig. 5) exhibited markedly different dynamics, maintaining over 500 linked pairs from April 2020 through December 2020. This number then gradually declined to approximately 300 pairs by mid-2021, followed by stabilization between 350 and 400 linked pairs through late 2021.

Linked pair dynamics for the Omicron variant from April 2020 through late 2021.
These temporal patterns reflect the evolutionary trajectories of each variant during their respective periods of emergence and dominance. The sustained high number of epistatic interactions in Omicron compared with Alpha and Delta suggests a fundamentally different evolutionary strategy, potentially reflecting adaptation to widespread population immunity.
A key advantage of our HMM-based approach over previous prefix-based methods is the ability to classify linkage patterns based on their temporal dynamics. We identified three distinct patterns:
The vast majority (98.7%) of all possible position pairs remained unlinked throughout the study period, underscoring the biological significance of the detected linkages as nonrandom associations likely under functional selection.
Our focused analysis of the 45 possible pairs formed by the 10 key Alpha variant positions revealed a strikingly different distribution: 7% exhibited permanent linkage patterns, 11% showed transient dynamics, 60% displayed oscillating patterns, and 22% remained unlinked (Table 2). The significantly higher proportion of linked pairs among Alpha variant positions compared with all possible spike protein pairs (78% vs. 1.3%) is consistent with the variant’s successful emergence and spread during the study period. This supports our hypothesis that epistatic interactions contribute to viral fitness advantages and sustainable transmission. The predominance of oscillating linkages (60%) among Alpha variant positions suggests a complex evolutionary landscape with potentially competing selective pressures acting on specific mutation combinations, as visualized in Figure 2.
Distribution of Epistatic Relationship Patterns across SARS-CoV-2 Variants of Concern
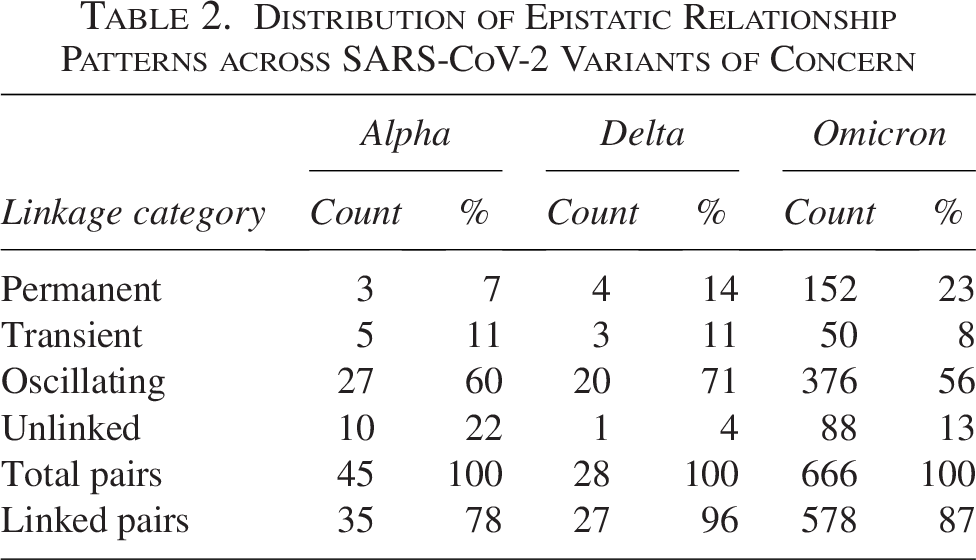
The temporal classification of epistatic linkages looks into how SARS-CoV-2 variants achieve evolutionary success. The striking enrichment of linked pairs among Alpha variant positions, 78% compared with just 1.3% when analyzing all 809,628 possible spike protein position pairs, demonstrates that VOC do not arise through random accumulation of mutations, but rather through the establishment of coordinated epistatic networks. The predominance of oscillating linkages (60%) among these Alpha variant pairs suggests that viral fitness landscapes are more dynamic than previously appreciated, with epistatic relationships responding to changing selective pressures throughout the variant’s emergence and spread.
This temporal perspective addresses a critical limitation of existing approaches (Mohebbi et al., 2023, 2024) for detecting epistatic interactions in SARS-CoV-2. While previous prefix-based methods successfully identified dense communities of linked mutations, they analyzed data across entire sampling periods, potentially obscuring the dynamic nature of these relationships. By modeling the temporal evolution of linkage states between SAVs, our HMM-based approach enables precise identification of when epistatic relationships form, dissolve, or fluctuate over time.
The three categories of temporal dynamics we identified, permanent, transient, and oscillating, each carry distinct evolutionary implications. The rarity of permanent linkages (only 0.3% of all possible SAV pairs across the spike protein) in our comprehensive analysis underscores their biological significance. These stable epistatic relationships likely represent mutation combinations under strong positive selection that, once established, provide consistent fitness advantages. Among Alpha variant positions, however, permanent linkages comprised only 7% of pairs, with oscillating patterns dominating at 60%. This suggests that even within successful variants, most epistatic relationships remain flexible, potentially allowing rapid adaptation to changing host immune landscapes.
The transient linkage category, while rare in our dataset (0.3% of total pairs), represents an especially intriguing class of epistatic interactions. These temporary associations might indicate mutations that become beneficial only under specific epidemiological conditions, such as changes in population immunity or transmission dynamic, before reverting to an unlinked state. The 11% of Alpha variant pairs showing transient patterns could reflect context-dependent advantages during specific phases of the variant’s emergence. Identifying these relationships could be crucial for understanding how SARS-CoV-2 adapts to shifting selective pressures.
Our approach demonstrated high sensitivity, successfully detecting all 17 previously identified epistatic pairs from prefix-based approaches (Mohebbi et al., 2024) while discovering an additional 18 significant linkages. Several of these newly identified relationships involve position 69 (part of the characteristic 69–70 deletion in Alpha) connecting with positions 614, 681, and 982, mutations known to impact spike protein conformation and function. These connections were likely missed by previous methods due to their inability to detect relationships that emerged later in the sampling period.
The temporal order noise reduction approach employed in our methodology enhances the robustness of our findings, with a stringent threshold (>80% agreement across 5 permutations) ensuring that identified linkages represent genuine biological signals rather than stochastic noise. This aspect of our methodology addresses a common challenge in epistasis detection: distinguishing biologically meaningful interactions from random associations.
A key limitation of our current implementation is the computational challenge associated with the exhaustive pairwise analysis of large genomic datasets. While our parallelized implementation significantly improves efficiency, further optimizations and sampling strategies may be necessary for real-time surveillance applications analyzing millions of sequences. Additionally, extending the model to detect higher-order epistatic interactions (beyond pairwise) represents an important direction for future research. A tractable approach could involve a hierarchical strategy: first identifying strongly linked pairs, then testing for additional positions that show coordinated patterns with established pairs, building interaction networks incrementally. This would be computationally feasible while capturing key multi-site interactions that likely contribute to complex viral phenotypes.
Future work could explore sampling strategies and approximate inference methods Stern et al. (2019) that prioritize analysis of positions with higher mutation frequencies, potentially reducing computation by 60%–70% without significant loss of sensitivity. GPU acceleration and intelligent heuristics could further enable real-time surveillance applications.
Our analysis of Delta and Omicron variants revealed distinct epistatic patterns that warrant further investigation. Delta showed 96% linked pairs with predominantly oscillating dynamics (71%), while Omicron exhibited 87% linkage with a more balanced distribution between permanent (23%) and oscillating (56%) patterns. These variant-specific patterns could reflect different evolutionary strategies—Delta’s high proportion of oscillating linkages suggesting rapid adaptation, while Omicron’s increased permanent linkages might indicate a more stable fitness peak achieved through extensive epistatic coordination among its 37 characteristic mutations.
Our binary encoding of mutations (present/absent) was chosen for computational tractability and interpretability. However, extending to the full amino acid alphabet could reveal additional epistatic patterns specific to particular amino acid substitutions. Such an extension would require a 400-state observation space per position pair and substantially larger datasets, but could provide insights into the biochemical basis of epistatic interactions.
Several limitations should be considered when interpreting our results. The fixed emission probabilities based on theoretical linkage disequilibrium, while providing computational efficiency and interpretability, may overlook complex mutation frequency biases. Future implementations could explore adaptive or Bayesian approaches that refine these probabilities based on observed data patterns. Additionally, our analysis remains sensitive to sequencing data quality and sampling biases. Although our temporal aggregation and consensus approach partially mitigate these issues, subtle dynamic linkages may be missed due to uneven temporal coverage. The detected linkages likely represent the most robust signals in the data.
In conclusion, our HMM-based approach represents a significant advancement in the detection and characterization of epistatic interactions in viral genomes. By capturing the temporal dynamics of linkage formation, this approach provides a more nuanced understanding of how epistatic networks evolve during variant emergence. The methodological principles developed here extend beyond SARS-CoV-2 surveillance, offering a general framework for studying epistasis in any rapidly evolving pathogen where temporal dynamics play a crucial role in adaptation.
AUTHORS’ CONTRIBUTIONS
A.E.A. developed and implemented the algorithms, performed data analysis, and wrote the paper. A.Z. developed the algorithms, designed the research, and supervised the project. P.S. and A.J. provided the data and performed data analysis. M.P. performed data analysis and wrote the article.
Footnotes
ACKNOWLEDGMENT
The authors gratefully acknowledge all data contributors, that is, the authors and their originating laboratories responsible for obtaining the specimens, and their submitting laboratories for generating the genetic sequence and metadata and sharing via the GISAID Initiative, on which this research is based.
AUTHOR DISCLOSURE STATEMENT
The authors declare that they have no conflicting financial interests.
FUNDING INFORMATION
A.Z. was supported by NSF grants 2212508 and 2316223.