
Abstract
Protein O-linked threonine glycosylation (OTG) is a crucial post-translational modification in eukaryotic species, playing a vital role in diverse biological processes. In humans, dysregulation of OTG has been associated with serious diseases, including cancer and neurological disorders. However, experimental detection of OTG sites remains costly and labor-intensive, underscoring the need for effective computational approaches. In this study, we introduce DP-OTG, a feature-free deep learning model for the accurate prediction of human OTG sites. Unlike existing tools that rely heavily on handcrafted features or large-scale pretrained language models, DP-OTG employs a hybrid architecture combining multi-kernel convolutional neural networks, bidirectional long short-term memory, and a trainable embedding layer to automatically learn sequence patterns directly from raw protein sequences. This end-to-end framework captures both local and long-range sequence dependencies without the need for manual feature engineering. Extensive evaluations using 10-fold cross-validation and independent testing demonstrate that DP-OTG achieves superior predictive performance, with an accuracy of 88.8% and an Matthew’s Correlation Coefficient (MCC) of 0.776 on the balanced test set, and an accuracy of 89.3% and an MCC of 0.661 on the imbalanced test set, outperforming several state-of-the-art predictors. In addition, to comprehensively assess the discriminative power and generalization ability of DP-OTG in predicting human OTG sites, we employed t-distributed stochastic neighbor embedding to visualize the feature representations before and after training. These results underscore the effectiveness of DP-OTG in extracting robust features for accurate OTG site prediction, even under challenging data distributions. Our findings highlight DP-OTG as a robust, efficient, and scalable tool for human OTG site prediction. All the code and resources related to this study have been made freely accessible at: https://github.com/nuinvtnu/DP-OTG/.
Keywords
1. INTRODUCTION
Glycosylation is a complex post-translational modification (PTM) that plays a vital role in protein folding, stability, and cellular communication (Hart, 1992; Haltiwanger and Lowe, 2004; Arey, 2012; Jayaprakash and Surolia, 2017). It exists in two primary forms: N-linked and O-linked glycosylation. Unlike N-linked glycosylation, O-linked glycosylation lacks a conserved sequence motif, making its site-specific prediction particularly challenging (Lis and Sharon, 1993). Notably, glycosylation has been observed in human cancer cells for decades and has also been implicated in Alzheimer’s disease, highlighting its potential significance in disease mechanisms (Schedin‐Weiss et al., 2014; Oliveira-Ferrer et al., 2017; Reily et al., 2019; Magalhães et al., 2021). However, experimental methods for identifying O-glycosylation sites are labor-intensive and costly, underscoring the need for efficient computational models.
Several machine learning-based methods have been developed to predict O-linked glycosylation sites, such as GlycoMine (Li et al., 2015), GlycoEP (Chauhan et al., 2013), OGP (Huang et al., 2021), and NetOGlyc-4.0 (Steentoft et al., 2013). These tools leverage various feature extraction techniques and classification algorithms to identify glycosylation sites with different levels of accuracy and efficiency. For instance, GlycoMine integrates multiple sequence- and structure-based features, while GlycoEP employs an ensemble approach to enhance predictive performance. OGP utilizes physicochemical properties and evolutionary information, whereas NetOGlyc-4.0 applies artificial neural networks to capture complex sequence patterns. Despite their effectiveness, many of these methods rely on handcrafted feature engineering, which can be time-consuming and computationally expensive. Moreover, their generalizability across different datasets remains a challenge, highlighting the need for more robust and efficient predictive models.
Notably, four recent tools, O-GlyThr (Tang et al., 2023), HOTGpred (Pham et al., 2024), Stack-OglyPred-PLM (Pakhrin et al., 2024), and DOGpred (Lee et al., 2025), have been proposed for O-GlyThr site prediction. O-GlyThr utilized seven handcrafted features combined with traditional classifiers like Random Forest. More recent approaches have integrated pretrained protein language models (PLMs) to extract informative sequence embeddings. HOTGpred, published in 2024, employs a handcrafted feature selection strategy, integrating 25 features, including 14 PLM-based embeddings (e.g., CPCProt, ProtTransXLNetUniRef100, Word2Vec), and 11 conventional descriptors (e.g., AAIndex1, composition-transition-distribution descriptors). Additionally, it applies machine learning models to refine feature selection, resulting in a high-dimensional representation (4864 features), and utilizes the XGBoost algorithm for classification. Stack-OglyPred-PLM, published in 2024, utilizes the ProtT5-XL-UniRef50 protein language model to generate contextualized embeddings for sites of interest (“S/T”), which are then fed into a Meta-Ensemble Model. A stacked generalization (meta-ensemble) approach is applied, where two Multi-Layer Perceptron models trained on ProtT5 and Ankh PLM embeddings are combined using a meta-model to optimize prediction accuracy. Meanwhile, DOGpred, published in 2025, follows a similar approach but with a more compact feature set of 18 features, comprising 9 conventional descriptors (e.g., AAIndex, Binary) and 9 embeddings from pretrained models (e.g., ESB, ESM, PTAB). DOGpred integrates a hybrid deep learning model combining convolutional neural network (CNN) and bidirectional long short-term memory (Bi-LSTM), offering an alternative predictive framework.
However, the above methods rely on manual feature engineering, which introduces subjectivity, demands extensive human effort, and separates feature extraction from model construction. Additionally, these tools require substantial computational resources for feature extraction, as they utilize large-scale PLMs, leading to long processing times. Despite advancements, existing methods still depend on manual feature selection, potentially limiting their ability to capture intricate sequence patterns comprehensively. Building on our previous research on PTMs and the development of hybrid deep learning models (Nguyen et al., 2017; Kao et al., 2020; Tran et al., 2023; Nguyen et al., 2024; Tran et al., 2025a), we aim to enhance and achieve more significant advancements in the prediction of protein O-linked Threonine Glycosylation (OTG) sites. In this study, we introduce DP-OTG, a feature-free deep learning model designed for accurate prediction of human OTG sites. This novel end-to-end architecture integrates deep learning with natural language processing (NLP) techniques, eliminating the need for handcrafted features. Our approach leverages a combination of multi-kernel 1D-CNN, Bi-LSTM networks, and NLP-based sequence encoding to automatically capture both local patterns and long-range dependencies within protein sequences. This integrated design enables the model to effectively identify diverse glycosylation-related motifs across multiple scales while maintaining the semantic integrity of the sequences.
Through extensive experiments, we demonstrate that our proposed model outperforms state-of-the-art methods on both balanced and imbalanced datasets, highlighting its effectiveness and robustness in O-glycosylation site prediction.
2. METHODS
Data collection and preprocessing
In this study, the datasets of human OTG were collected from HOTGpred (Pham et al., 2024), DOGpred (Lee et al., 2025), O-GlyThr (Tang et al., 2023), UniProt database (UniProt Consortium, 2024), and relative literatures. After some technical steps to remove redundant data, we decided to utilize the same dataset as the most recent studies used on human OTG sites prediction, including HOTGpred (Pham et al., 2024), DOGpred (Lee et al., 2025), and O-GlyThr (Tang et al., 2023). This resulted in 318 human OTG proteins. The decision to adopt this dataset in our study was driven by several key factors. First, using the same dataset as previous state-of-the-art models ensures a fair and direct comparison of model performance. Second, its widespread adoption establishes it as a standard reference for evaluating new models. Lastly, the inclusion of an imbalanced test set allows for a more realistic assessment of the model’s ability to handle scenarios in which human non-OTG sites significantly outnumber human OTG sites—an essential consideration for practical applications. By leveraging this dataset, we ensure consistency in evaluation and provide meaningful comparisons with the latest advancements in OTG prediction.
To remove duplicate and redundant proteins, the CD-HIT tool (Km et al., 2020) was applied with a 40% sequence identity threshold, which refined the dataset to 246 unique human proteins. Since this study focuses on the sequence-based characterization of threonine (T) sites and their substrate specificities, extracting sequence fragments from the full FASTA sequence of proteins is necessary. It is worth noting that this study specifically focuses on O-linked glycosylation occurring at threonine (Thr) residues. This decision was made to ensure consistency with previous benchmark datasets (HOTGpred, DOGpred, and O-GlyThr), which were also designed exclusively for threonine-based glycosylation prediction. Furthermore, the number of experimentally verified Thr sites in humans is considerably higher and more reliable than Ser sites, providing a stronger foundation for model training and evaluation. Future studies will aim to extend this framework to include serine and mixed O-linked glycosylation sites for a more comprehensive analysis. Consistent with previous research, positive samples were constructed by extracting sequence fragments using a window size of
Based on preliminary technical steps to investigate with different window sizes, the optimal window size was found to be 41; therefore, we decided to utilized window size of 41 to extract peptides from full FASTA protein sequences for this study. For negative samples, threonine (T) residue not experimentally verified as OTG sites were treated as non-OTG sites, and the same window size of
Training Dataset and Testing Dataset to Use in This Study

Training Dataset and Testing Dataset to Use in This Study
Through the process of extracting peptides and removing homologous data, the final dataset consisted of 1078 positive and 110,923 negative samples. To mitigate class imbalance, 1078 negative samples were randomly selected to balance the dataset, forming a training set of 878 positive and 878 negative samples, along with a balanced test set of 200 positive and 200 negative samples. Additionally, an imbalanced independent test set, introduced by Pham, N.T et al. (Pham et al., 2024), extends the balanced test set by incorporating 800 additional negative samples randomly selected from the original negative pool, reflecting real-world data distribution.
Similar to human natural language, protein sequences can be naturally represented as character strings. The protein alphabet consists of 20 common amino acids, excluding rare and uncommon ones. Like natural language, naturally evolved proteins exhibit modular structures with slight variations, arranged hierarchically. In this analogy, common protein motifs and domains, which serve as fundamental functional units, resemble words, phrases, and sentences in human language. This structural similarity makes NLP-based encoding a powerful approach for protein sequence analysis (Ofer et al., 2021).
In this study, protein sequences are first tokenized using the 1-gram technique, where each amino acid is treated as an individual unit. Each amino acid is subsequently assigned an integer index based on a predefined amino acid dictionary before being passed to the embedding layer. This choice ensures that the encoding fully preserves sequence order and primary structure information without introducing artificial dependencies. The tokenized sequences are then processed through the Embedding layer of a deep learning model, which maps each amino acid to a continuous vector space, enabling the model to capture biological relevance directly from data.
Unlike manual feature extraction methods that rely on predefined biochemical properties, this approach allows the model to learn adaptive representations through training. The Embedding Layer functions as a dynamic lookup table, refining vector representations as training progresses. Through forward propagation, loss computation, and backpropagation, embeddings evolve to capture sequence patterns critical for PTM site prediction. This integration of deep learning with NLP-based techniques enables automatic extraction of key sequence features, enhancing predictive performance without the need for handcrafted rules. The process of feature encoding using NLP techniques (Fig. 1) is summarized in two main steps as follows:
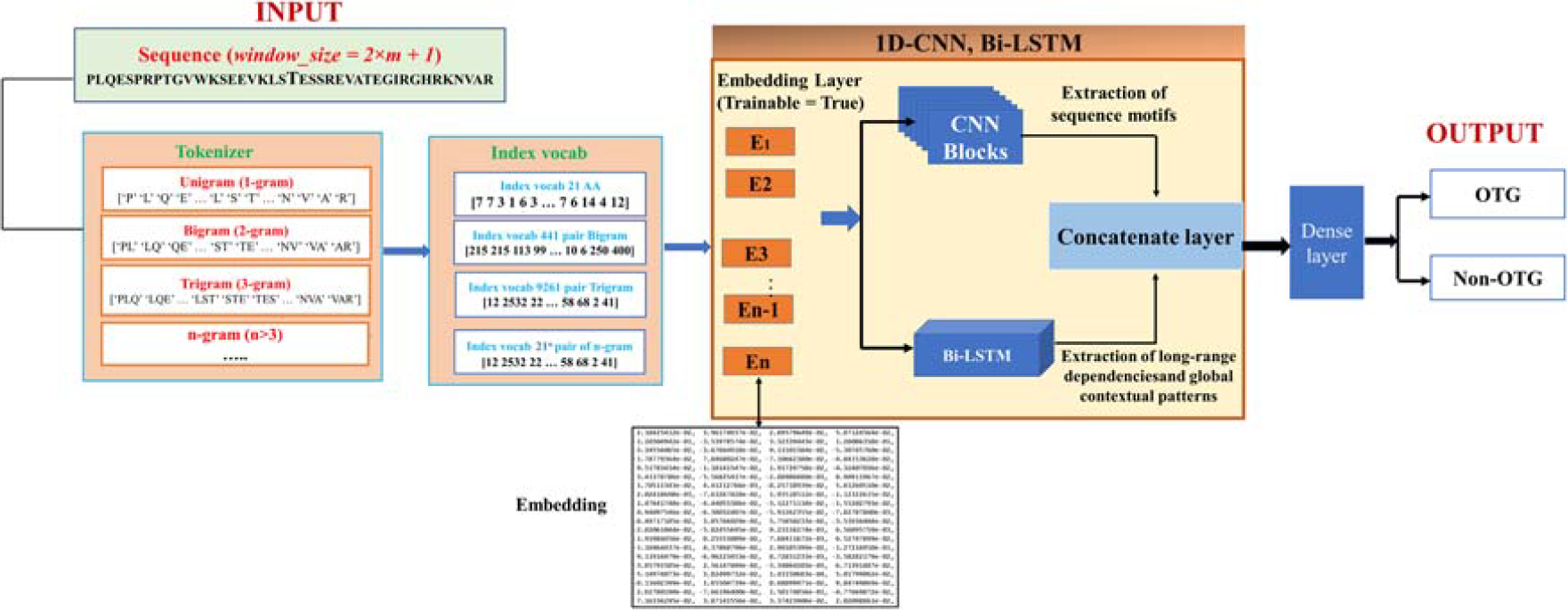
Illustration of the n-gram–based tokenization and embedding process used in the DP-OTG model.
To illustrate the tokenization and embedding procedure, Figure 1 presents an overview of the n-gram-based encoding concept, in which protein sequences can theoretically be decomposed into overlapping residue fragments (e.g., 1-gram, 2-gram, or 3-gram). Each token is then converted into a numerical index based on a predefined amino acid vocabulary and subsequently mapped to a dense vector through the Embedding layer. Although various n-gram examples are depicted to clarify the general principle, in this study, we employed only the 1-gram representation, as it provided the most informative and stable features according to our previous comparative experiments (Nguyen et al., 2024; Tran et al., 2025a, 2025b).
In an attempt to accurately and effectively predict human OTG sites, the hybrid deep learning architecture, named DP-OTG, has been designed. Our approach combines a five-layer 1 D-CNN, operating in parallel with three different kernel sizes, and a Bi-LSTM network, leveraging NLP-based word embedding techniques to automatically learn meaningful sequence representations from raw protein sequences. As depicted in Figure 2, the proposed model consists of three main parts: data collection and pre-processing; data encoding using NLP techniques; and network architecture.
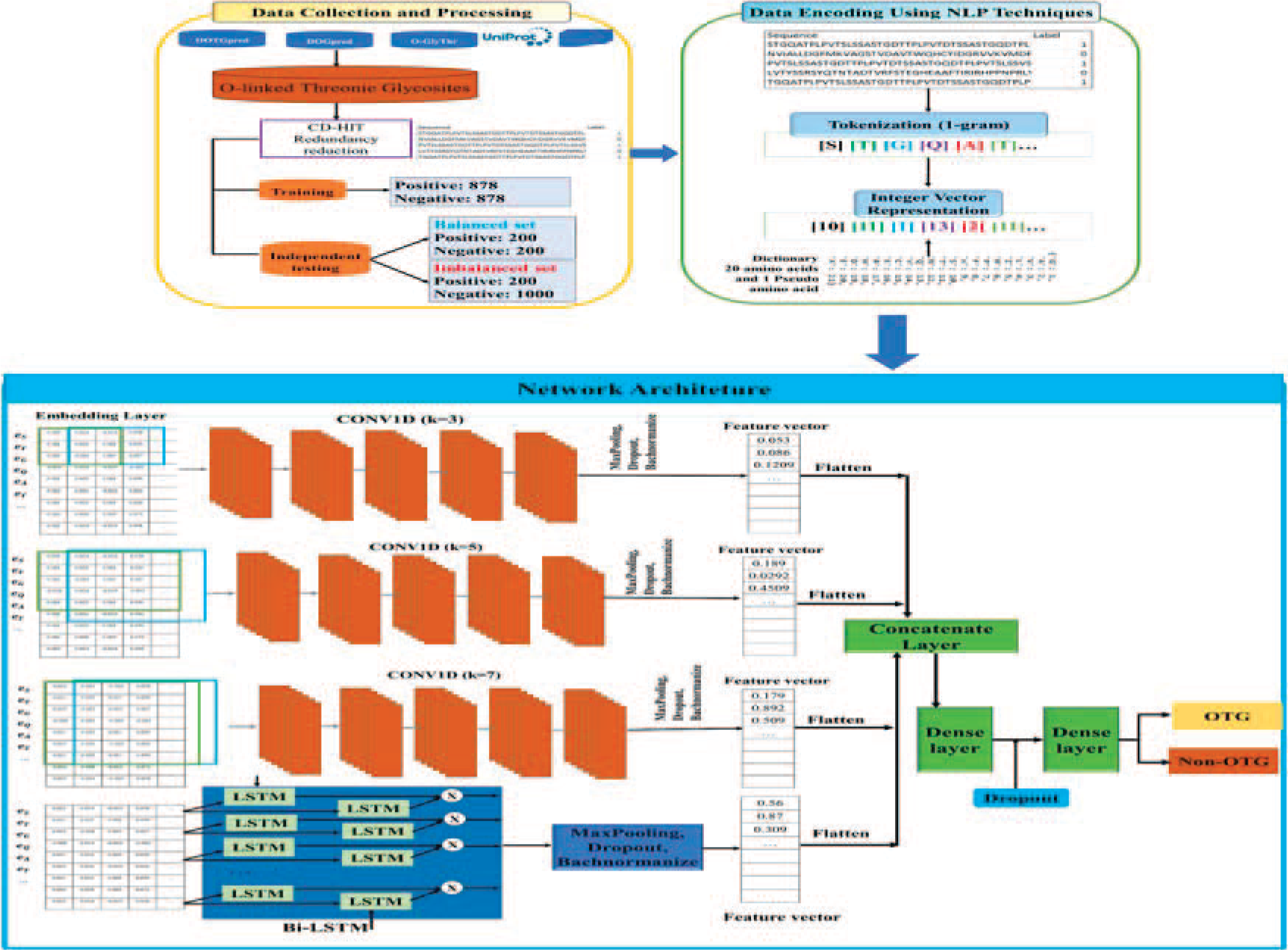
The system architecture of the proposed model.
The model consists of three parallel CNN branches, each using a different kernel size (3, 5, and 7) to extract features at different levels. Each CNN branch includes five Conv1D layers with 32 filters, ReLU activation, and “same” padding to preserve the output size. After the convolutional layers, each branch applies a MaxPooling1D layer with a pool size of 2 to reduce dimensionality, followed by a Dropout layer (rate of 0.4) to prevent overfitting, and a BatchNormalization layer to normalize activations. In addition to the CNN branches, the model includes a Bi-LSTM branch, which applies a Bi-LSTM layer with 32 hidden units to the embedded sequence, capturing contextual information in both forward and backward directions. This branch also incorporates a Dropout layer (rate of 0.4) and a BatchNormalization layer for improved training stability. The outputs from the three CNN branches and the Bi-LSTM branch are flattened using Flatten layers and then merged using a Concatenate layer. The fully connected network consists of two Dense layers with 128 and 64 units, respectively, both using ReLU activation and L2 regularization (coefficient 0.01) to mitigate overfitting. Each dense layer is followed by a Dropout layer with a dropout rate of 0.5. Finally, the output layer consists of 2 units with a softmax activation function, enabling the model to perform binary classification. The model uses a standard decision boundary of 0.5, where the class with a predicted probability greater than 0.5 is assigned as the positive class.
To provide a fair and comprehensive comparison, a sequential baseline model was additionally constructed. In this baseline architecture, three parallel branches were designed, each consisting of a 1D-CNN module with a kernel size of 3, 5, or 7, followed directly by a Bi-LSTM layer (CNN3 → Bi-LSTM, CNN5 → Bi-LSTM, and CNN7 → Bi-LSTM). Each CNN module shares the same configuration as the CNN branches in DP-OTG, including five Conv1D layers, max-pooling, dropout, and batch normalization. The Bi-LSTM layer in each branch contains 32 hidden units to capture long-range dependencies after kernel-specific convolutional feature extraction. The output features from the three sequential branches are then flattened and concatenated, followed by two fully connected Dense layers with 128 and 64 units, respectively, before the final softmax output layer. This sequential design serves as a baseline to investigate the impact of learning temporal dependencies after convolutional feature extraction, in contrast to the parallel feature learning strategy adopted in DP-OTG.
By integrating deep learning with NLP-based feature extraction, DP-OTG establishes a new benchmark for glycosylation site prediction, outperforming existing tools while maintaining computational efficiency. The core components of DP-OTG, along with its parameters and configurations, are summarized as in Table 2.
The Information of DP-OTG’s Components and Its Parameters

The operation and prediction mechanism of the proposed model is carried out in four steps: (1) Create 1-gram dictionary; (2) Sequences encoding; (3) Train and validate the predictive model; and (4) Make predictions and get predicted outputs. Detailed information is illustrated in Algorithm DP-OTG, as shown in Figure 3.
Algorithm DP-OTG: Accurate prediction of human OTG sites.
The overall architecture and operational mechanism of the proposed model confer significant advantages, enabling it to outperform existing prediction tools for human OTG site identification. The key advantages of the DP-OTG model are summarized as follows:
Automatic Feature Extraction: Unlike traditional approaches that depend on handcrafted features, DP-OTG automatically learns sequence patterns directly from raw input data. Trainable Word Embeddings: The embedding layer is optimized during training, enabling the model to learn task-specific representations for human OTG sites prediction. Hybrid Learning Strategy: By combining 1D-CNN to capture local sequence motifs and Bi-LSTM to model long-range dependencies, the model achieves a more comprehensive and robust representation of protein sequences. Computational Efficiency: In contrast to large-scale PLMs, DP-OTG model provides a lightweight and scalable solution, making it well-suited for large-scale PTM prediction tasks.
In order to evaluate the performance of the predictive models, the 10-fold cross-validation approach has been performed to assess the classifying power of the predictive models. The following measurements are commonly used to evaluate the performance of the constructed models: Sensitivity (SEN), Specificity (SPE), Accuracy (ACC), Matthew’s Correlation Coefficient (MCC), l, and F1-score, area under the curve (AUC).






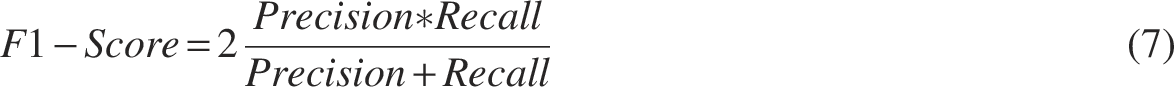
Herein, TP, TN, FP, and FN represent the numbers of true positives, true negatives, false positives, and false negatives, respectively.
After running a 10-fold cross-validation process, the predictive model with the highest MCC and accuracy values is selected as the optimal model for identifying human OTG sites. Additionally, an independent testing approach was conducted to evaluate the selected model’s performance in a real-case scenario. Furthermore, a comparison between our proposed model and a recent relevant predictor of human OTG sites was performed to assess the practical effectiveness of the proposed model.
3. RESULTS
To examine the position-specific amino acid composition for Human OTG sites, WebLogo (Crooks et al., 2004) was applied to generate the graphical sequence logo, visualizing the relative frequency of amino acids at positions surrounding glycosylation sites. The sequence entropy plots in WebLogo clearly depicted the conserved motifs within the substrate sequences, allowing for the identification of amino acid preferences around glycosylation sites.
The comparative analysis of amino acid composition between human OTG sites (positive data) and non-OTG (negative data) revealed notable differences. As illustrated in Figure 4A, the most prominent amino acids in human OTG sites included Threonine (T), Serine (S), and Proline (P), while amino acids such as Tryptophan (W), Cysteine (C), and Methionine (M) were observed less frequently. Although Threonine (T), Serine (S), and Proline (P) appeared frequently in both positive and negative datasets, their occurrence was nearly twice as high in the positive dataset.

Frequency of the amino acid composition surrounding the human OTG sites:
To further distinguish glycosylation from non-glycosylation sequences, a TwoSampleLogo (Vacic et al., 2006) analysis was performed, as shown in Figure 2B. The enriched residues in glycosylation sites predominantly included Threonine (T), Proline (P), and Serine (S), while the depleted residues were primarily nonpolar amino acids such as Leucine (L), Glycine (G), and Valine (V). These findings provide valuable insights into the sequence characteristics of human OTG sites and contribute to the development of predictive models for identifying human OTG sites.
As summarized in Table 3, Figure 5 and Figure 6, the 10-fold cross-validation results demonstrate the effectiveness of different 1D-CNN models and Bi-LSTM in human OTG sites prediction. Various CNN architectures with different kernel sizes were evaluated based on key classification metrics, including Specificity (SPE), Sensitivity (SEN), Accuracy (ACC), MCC, and F1-score. To build an optimal predictive model, we conducted experiments on individual 1D-CNN models with different kernel sizes (3, 5, 7) as well as hybrid 1D-CNN architectures that combined these kernels. We identified the highest-performing hybrid 1D-CNN architecture and then integrated it with the Bi-LSTM network to capture a more comprehensive set of features from the data.
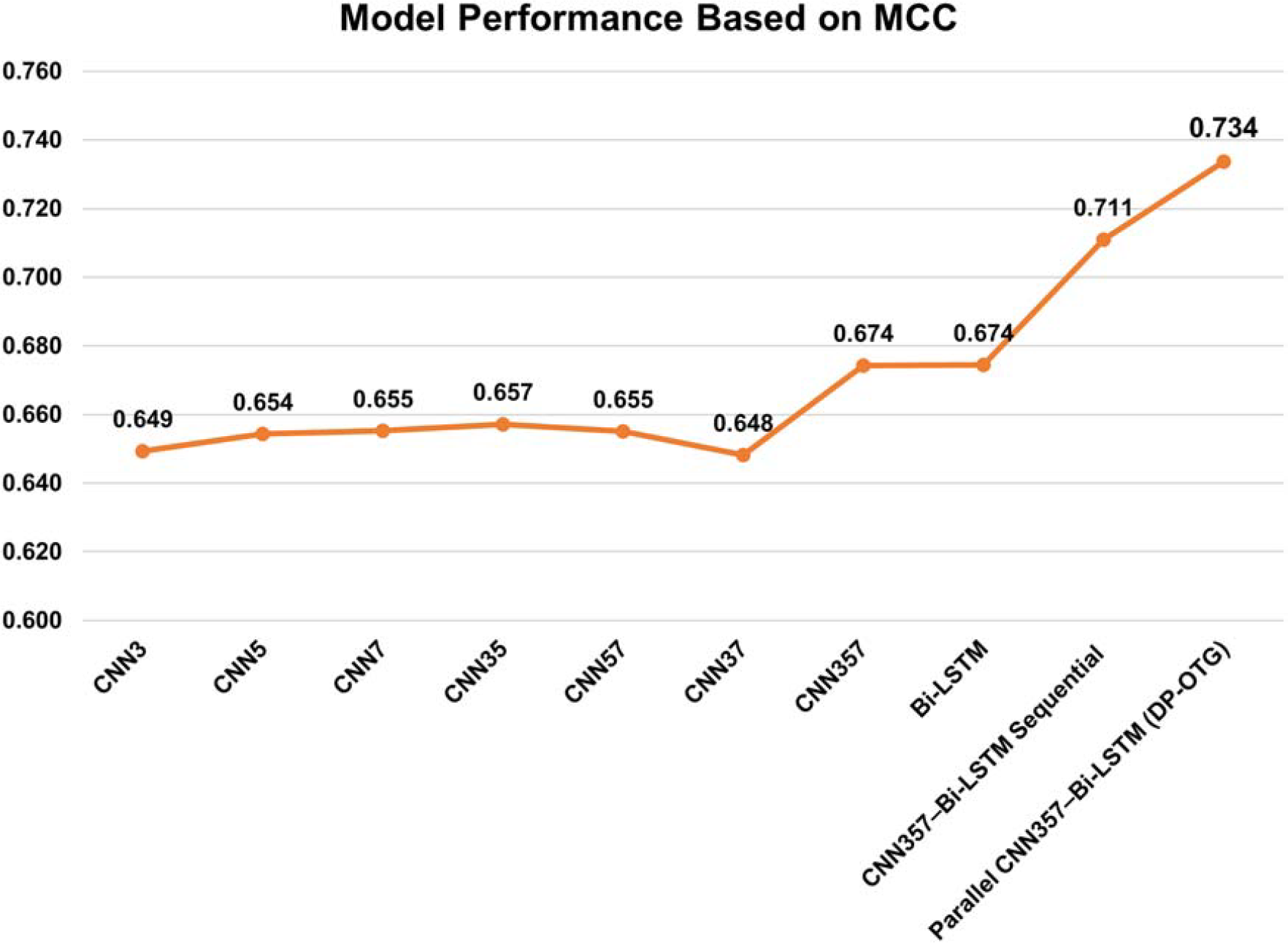
Comparison of MCC values across all predictive models for OTG site identification using 10-fold cross-validation. MCC, Matthew’s Correlation Coefficient.

The ROC curve of DP-OTG model evaluated by 10-fold cross-validation.
Performance Evaluation by 10-Fold Cross-Validation

Among the CNN-based models, CNN357, a hybrid 1D-CNN architecture incorporating three different kernel sizes, demonstrated superior performance. Each kernel in CNN357 learns distinct motifs from the data, enhancing the model’s ability to extract relevant sequence patterns. CNN357 achieved the highest accuracy (83.6%) and MCC (0.674), indicating its strong capability in feature extraction. The Bi-LSTM model, designed to capture long-range dependencies, exhibited balanced performance with an accuracy of 83.7% and an MCC of 0.674, further confirming its robustness in distinguishing glycosylation sites.
In addition to the above architectures, we further introduced a sequential baseline model to provide a more comprehensive comparison. In this baseline, three parallel branches were constructed, each consisting of a CNN layer with kernel sizes of 3, 5, and 7, followed by a Bi-LSTM layer (CNN3→Bi-LSTM, CNN5→Bi-LSTM, and CNN7→Bi-LSTM). The learned features from these three sequential branches were concatenated and subsequently passed through two fully connected dense layers to generate the final prediction. As reported in Table 3, this sequential CNN357–Bi-LSTM baseline achieved improved performance compared to individual CNN and Bi-LSTM models, with notable gains in accuracy (85.5%) and MCC (0.711), indicating that modeling long-range dependencies after kernel-specific convolutional feature extraction can enhance feature representation.
The parallel CNN357–Bi-LSTM (
Comparatively, the superior performance of the parallel DP-OTG model over the sequential baseline suggests that allowing CNN and Bi-LSTM components to learn complementary representations simultaneously is more effective than enforcing a strictly sequential dependency. This parallel design facilitates richer feature interactions between local sequence motifs and long-range contextual information, leading to more discriminative and stable predictions across cross-validation folds.
The relatively low standard deviations across all metrics indicate that the model’s performance is stable and not significantly affected by variations in the training and testing partitions. These findings confirm the generalization ability of the model in distinguishing between positive and negative samples in the context of glycosylation site prediction.
To further evaluate the classification capability of our model, we employed t-distributed Stochastic Neighbor Embedding (t-SNE) to visualize the feature representations of the training dataset before and after training. Prior to training, the data points corresponding to positive and negative samples were largely intermixed, indicating limited separability in the raw feature space. However, after training, the t-SNE projection revealed two clearly separated clusters, corresponding to the positive and negative classes. This distinct separation demonstrates that the model successfully learned discriminative features, leading to strong classification performance. The results confirm the model’s effectiveness in capturing meaningful representations that enhance glycosylation site prediction (Detail in Fig. 7).
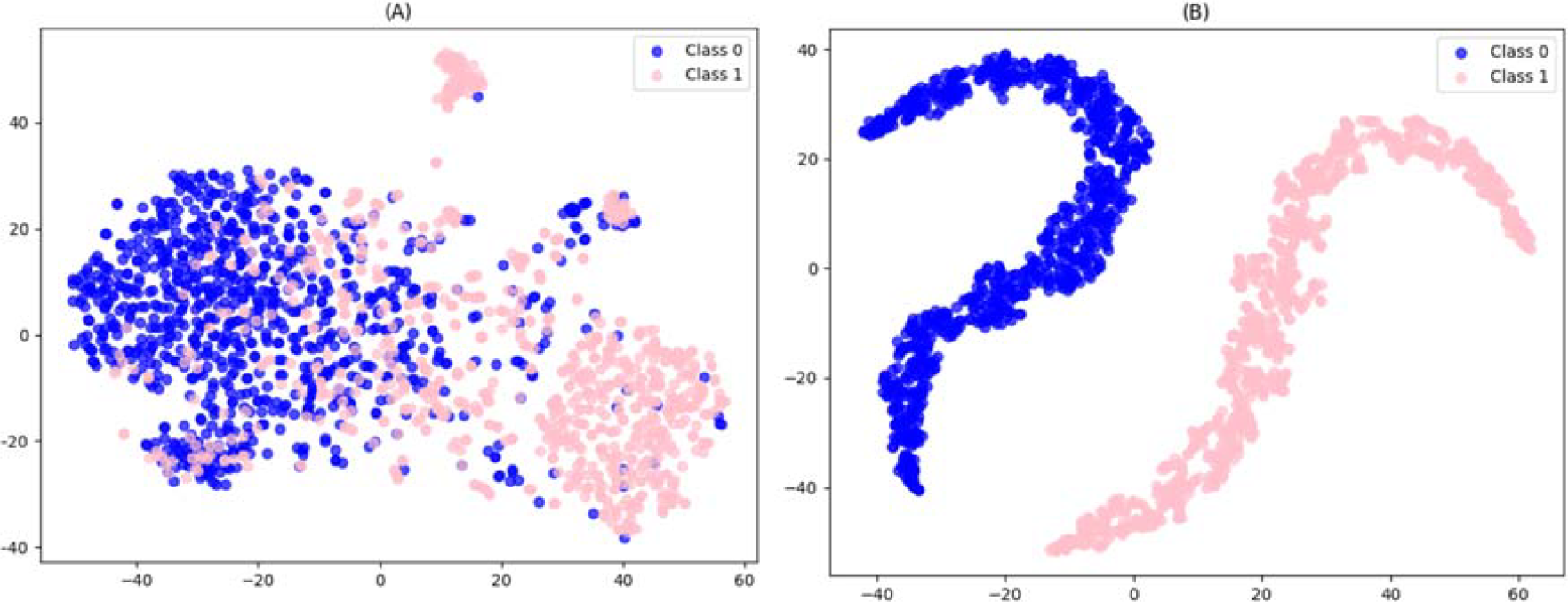
t-SNE visualization of feature representations before and after training.
To validate the effectiveness of our proposed model in real-world scenarios, we conducted independent testing on two distinct test sets: a Balanced test set and an Imbalanced test set. As summarized in Table 4, Figures 8 and 9, the results demonstrate the superior performance of the

Comparison of model performance based on MCC scores on two independent test sets: one balanced and one imbalanced.
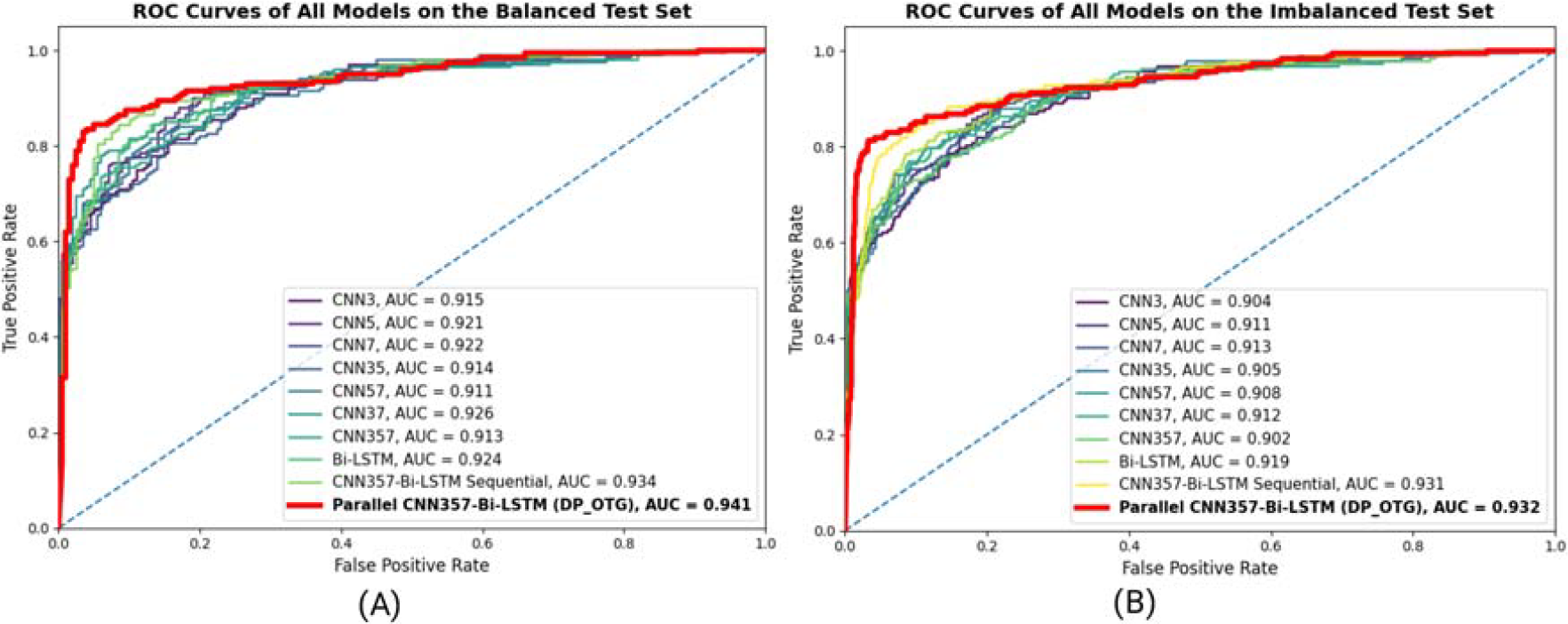
The ROC curves of predictive models on the Balanced test set and Imbalanced test set.
Performance Evaluation by Independent Testing (Imbalanced and Balanced Test)
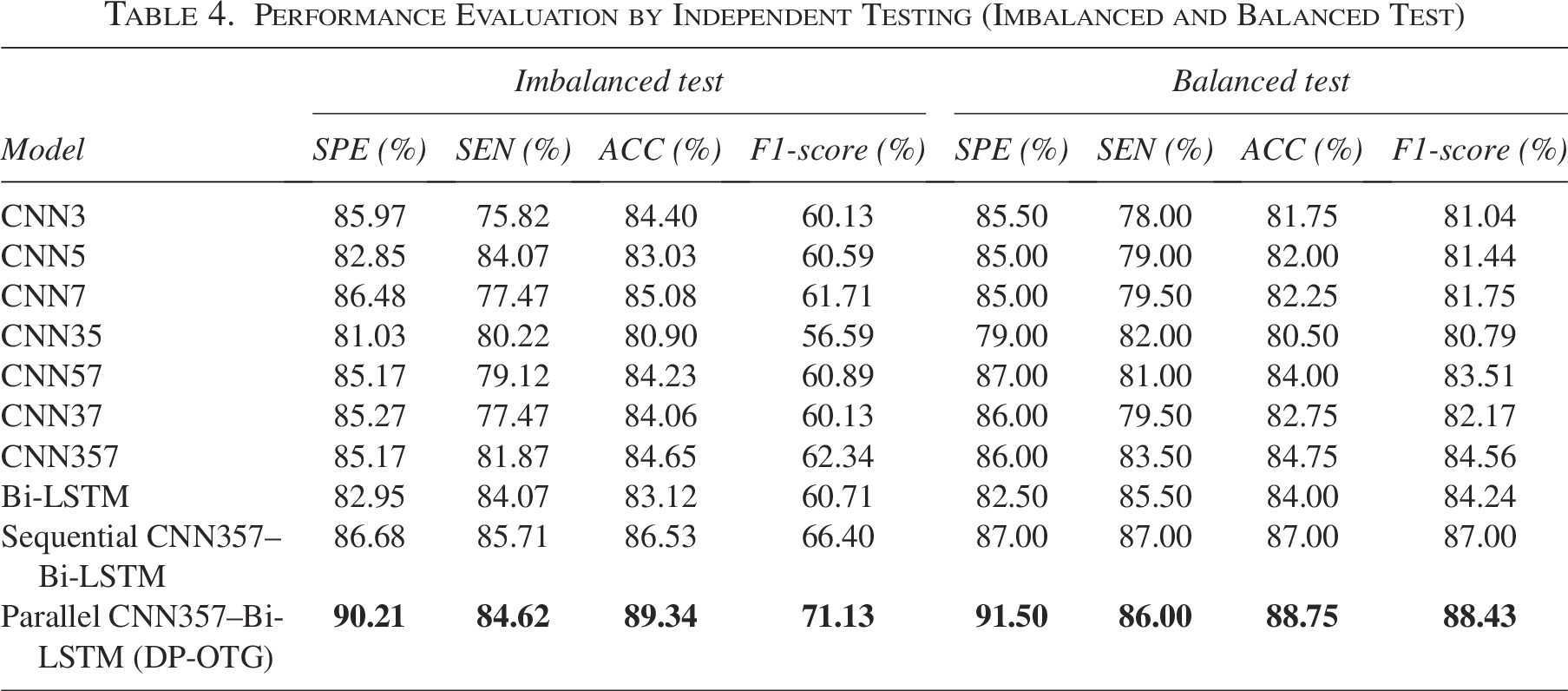
On the balanced test set,
Beyond overall accuracy and MCC, recall is a particularly critical metric for imbalanced datasets, as it directly reflects the model’s ability to correctly identify true glycosylation sites. To provide a more comprehensive evaluation, Precision–Recall (PR) curves were plotted for all models on both the balanced and imbalanced test sets, as illustrated in Figure 10. As shown by the recall curves on the imbalanced test set (Fig. 9), the DP-OTG model consistently maintains higher recall values across a wide range of decision thresholds compared with all baseline models. This indicates that DP-OTG is more effective at reducing false negatives, which is essential for practical glycosylation site prediction tasks. The proposed

Comparison of Precision–Recall curves among all models on the balanced and imbalanced test sets, highlighting the superior performance of the proposed parallel CNN357_Bi-LSTM (DP-OTG) model.
The strong recall and PR performance of DP-OTG on the imbalanced test set can be attributed to its parallel CNN–Bi-LSTM architecture, which effectively integrates multi-scale local feature extraction with long-range dependency modeling. Compared with individual CNN variants and the sequential CNN–Bi-LSTM baseline, DP-OTG achieves a more favorable recall–precision trade-off, enabling the detection of a larger proportion of true glycosylation sites without a substantial increase in false positive predictions.
Overall, these findings confirm that the
To evaluate the generalization capability of our model on unseen data, we applied t-SNE to visualize the feature representations of two independent test sets: one with a balanced distribution of positive (label 1) and negative (label 0) samples, and another with an imbalanced distribution. As illustrated in Figure 11(A) and Figure 12(A), in the raw feature space, the positive and negative samples are largely intermixed, indicating limited inherent separability prior to modeling. However, after passing the test data through the trained model, the transformed feature spaces show clearly separated clusters corresponding to the two classes, as displayed in Figure 11(B) and Figure 12(B). This distinct class separation demonstrates the model’s ability to extract discriminative and generalizable representations, enabling accurate classification of glycosylation sites even in scenarios involving class imbalance.

t-SNE visualization of balance test data.

t-SNE visualization of imbalanced test data.
To evaluate the effectiveness of our proposed DP-OTG model, we compared its performance against recent relevant prediction tools for human OTG using the same training dataset and an independent test dataset. The results are summarized in Table 5 and Figure 13, where both balanced and imbalanced test sets were used to assess each model’s robustness.
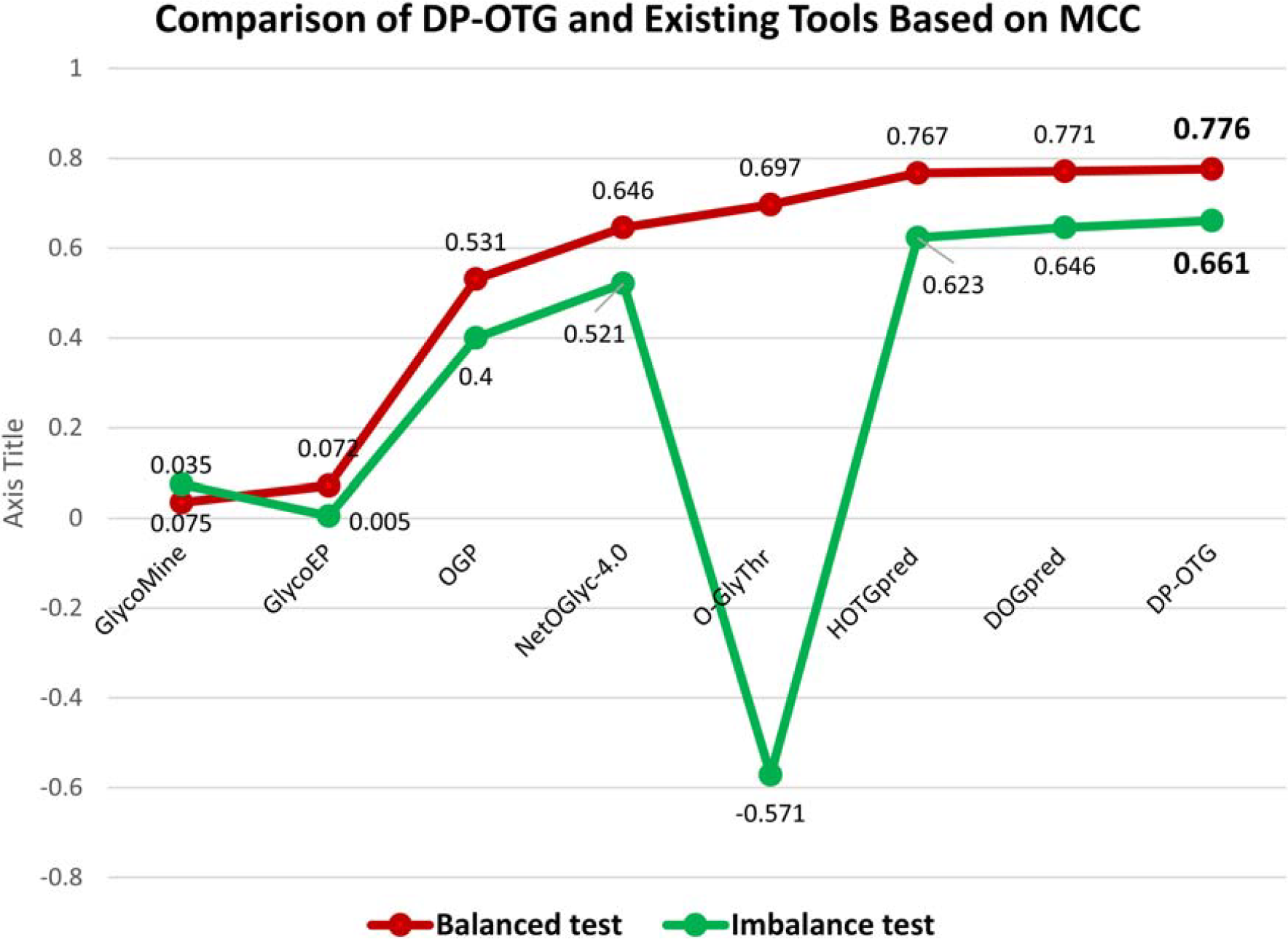
Comparison of DP-OTG and Existing Tools Based on MCC across Balanced and Imbalanced Tests.
Comparison of the Performance of the Proposed Model with Other Tools Using the Same Test Dataset
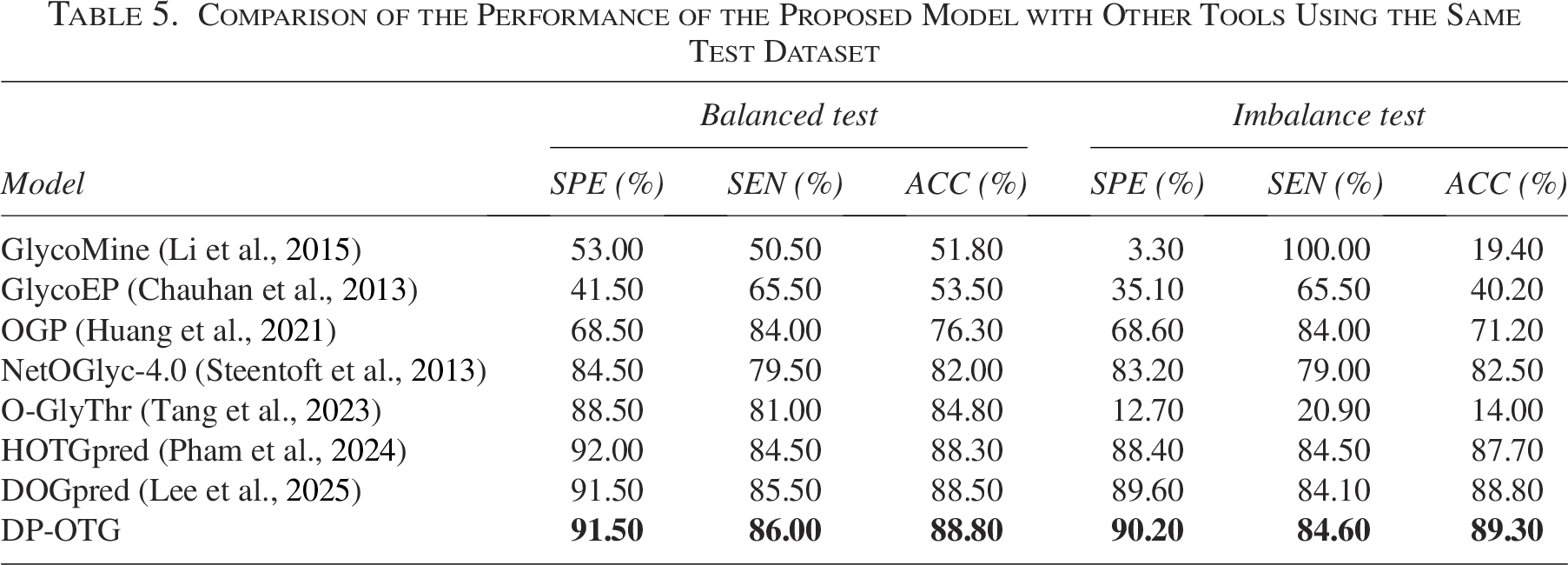
On the balanced test set, the DP-OTG achieves an ACC of 88.8%, an MCC of 0.776, and an AUC of 0.941, outperforming most existing models. While HOTGpred and DOGpred obtain slightly higher AUC values (0.943 and 0.947, respectively), DP-OTG maintains a better balance between sensitivity and specificity. Importantly, our model does not rely on handcrafted feature engineering or computationally expensive pretrained PLMs, making it a more efficient alternative.
For the imbalanced test set, DP-OTG demonstrates superior performance, achieving an ACC of 89.3%, an MCC of 0.661, and an AUC of 0.932. This highlights its robustness under real-world conditions, where class distributions are often skewed. In contrast, models such as O-GlyThr and GlycoEP experience substantial drops in performance, with negative MCC values and reduced predictive stability. Even state-of-the-art predictors like HOTGpred and DOGpred show a decline in MCC, whereas DP-OTG maintains consistent performance.
Overall, these results confirm that DP-OTG provides a strong balance between accuracy, computational efficiency, and generalizability. Unlike existing tools that either suffer from performance degradation on imbalanced data or require extensive feature extraction, DP-OTG delivers competitive performance with a streamlined architecture. This makes it a promising choice for large-scale applications requiring reliable PTM site prediction.
4. CONCLUSION
In this study, we proposed
Extensive cross-validation and independent testing confirm that DP-OTG surpasses existing predictors across both balanced and imbalanced datasets. As shown in Table 5, our model achieves the highest accuracy among all compared methods, with an ACC of 88.8% and an MCC of 0.776 on the balanced test set, and an ACC of 89.3% and an MCC of 0.661 on the imbalanced test set. These results demonstrate that DP-OTG not only improves predictive accuracy but also maintains strong performance under imbalanced conditions, where many existing tools experience performance degradation.
Our findings highlight the effectiveness of hybrid deep learning combined with NLP techniques in PTM site prediction. By eliminating manual feature selection while achieving superior accuracy. The ability to update word embedding vectors during training further enhances feature extraction, enabling the model to learn task-specific sequence representations dynamically.
AUTHORS’ CONTRIBUTIONS
T.-X.T.: Writing—original draft, methodology, investigation, formal analysis, data curation, and conceptualization. N.-Q.-K.L.: Methodology, writing—original draft, validation, supervision, methodology, and investigation. D.-H.L.: Methodology, writing—review and editing, methodology, and conceptualization. V.-N.N.: Writing—original draft, writing—review and editing, supervision, methodology, funding acquisition, data curation, and conceptualization.
Footnotes
ACKNOWLEDGMENT
The authors would like to express the sincerest gratitude to all the anonymous reviewers for their comments and opinions on the article, which were of great help to the article.
AUTHOR DISCLOSURE STATEMENT
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.
FUNDING INFORMATION
This work was funded by the Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 102.05-2023.49.
ETHICAL APPROVAL
This study does not involve human participants or animals and was therefore exempt from Institutional Review Board (IRB) approval.