
Abstract
RNA editing is a post-transcriptional modification that alters single-nucleotide sites within RNA strands, thus diversifying transcriptomes and proteomes and modulating gene expression. While better characterized in eukaryotes and in a few microbes, the study of RNA editing in entire microbiomes remains unexplored. Recent studies have demonstrated that A-to-I RNA editing contributes to bacterial adaptation and pathogenicity. Previously, we developed M
Here, we introduce ME
INTRODUCTION
RNA editing is an important post-transcriptional modification that alters RNA sequences, thereby expanding the functional diversity of the transcriptome (Gott and Emeson, 2000). In eukaryotes, RNA editing mechanisms (e.g., adenosine-to-inosine or A-to-I editing) catalyzed by adenosine deaminases (ADARs) are well-studied and known to influence diverse cellular processes, including neuronal development, immune responses, and disease pathogenesis (Blow et al., 2004; Christofi and Zaravinos, 2019; Mallela and Nishikura, 2012). In contrast, RNA editing in bacteria has not been investigated sufficiently. It was initially identified in bacterial tRNAs by Wolf et al. (2002) in the early 2000s. Until recently, editing in bacterial mRNAs was scarcely reported, with limited instances shown to influence bacterial pathogenicity and stress responses (Bar-Yaacov et al., 2017; Nie et al., 2020). RNA editing in the context of complex microbial ecosystems, such as the human gut microbiome, remains unexplored. The human gut microbiome comprises numerous bacterial taxa integral to host physiology, contributing to various health conditions including inflammatory bowel diseases (IBD). Understanding RNA editing within these microbial communities could reveal novel adaptive mechanisms critical for microbial survival and host–microbe interactions. Several computational tools have been developed to detect RNA editing in eukaryotes, including REDItools Picardi and Pesole (2013), RES-Scanner (Wang et al., 2016), and SPRINT (Zhang et al., 2017), which primarily rely on reference genomes and have been widely applied in eukaryotic systems. However, these approaches are not well-suited for microbiome-scale analyses, where incomplete reference catalogs, high strain-level diversity, and frequent horizontal gene transfer introduce substantial bias and limit sensitivity (Nayfach and Pollard, 2016; Lloyd-Price et al., 2019). To resolve this gap, we developed M
However, reliance on reference genomes limited the scope of M
METHODS
ME
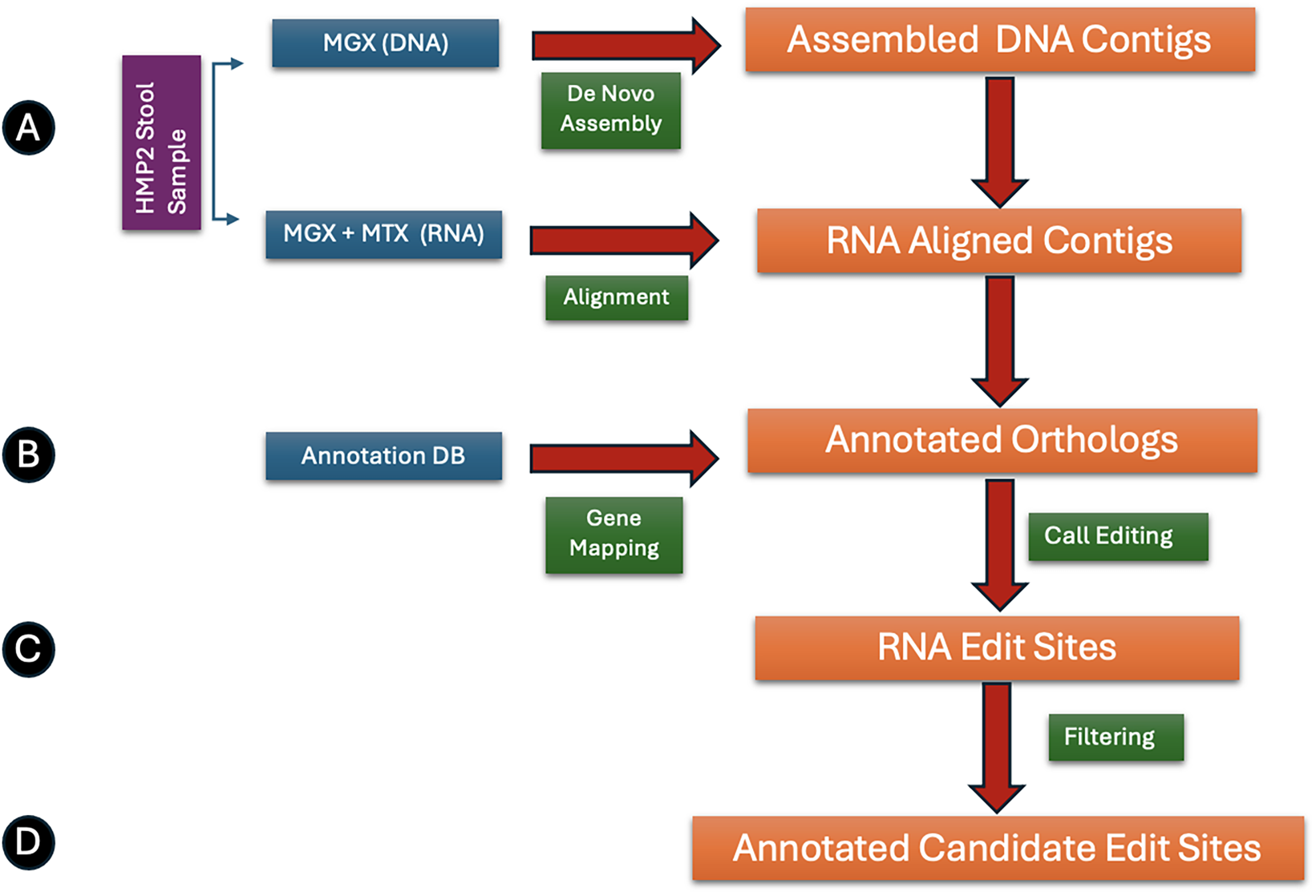
ME
Statistical methods are then applied to assess the significance of RNA editing at each site, followed by differential analysis between disease and healthy cohorts to identify edit sites potentially associated with pathophysiological states. Finally, functional annotation and orthology inference are carried out using eggNOG-mapper (Huerta-Cepas et al., 2019) to characterize the biological context of the edited loci, providing insights into their potential roles in microbial function and host–microbiome interactions.
Our dataset consisted of paired metagenomic and metatranscriptomic sequence datasets obtained from 834 samples from 109 individuals diagnosed with Crohn’s disease (CD), ulcerative colitis (UC), or non-inflammatory bowel disease (non-IBD) (Lloyd-Price et al., 2019), all of which were collected as part of the Integrative Human Microbiome Project (iHMP2). To minimize the confounding factors, the samples were screened to include samples exclusively from the Caucasian population, resulting in a final dataset of 748 paired samples from 96 individuals. To minimize population-specific genetic and environmental heterogeneity that could confound RNA editing analyses, we restricted this study to individuals self-reported as Caucasian, consistent with prior iHMP2 subgroup analyses. While this filtering improves internal consistency, it may limit generalizability and is addressed as a limitation

Quality control and data preprocessing
Raw sequencing reads underwent stringent quality control using Trim Galore (Krueger, 2012) and FastQC (Andrews, 2010) to remove adapters and low-quality bases (Phred score
De novo assembly
High-quality metagenomic reads were assembled into contigs using MEGAHIT (Li et al., 2015). Assembled contigs provide greater confidence for downstream steps such as annotations. It also allows for unbiased assemblies to be generated, suitable for diverse microbial communities. All metagenomic reads were then aligned to the assembled contigs using Bowtie2 (Langmead and Salzberg, 2012), followed by an identical step with metatranscriptomic reads. Algorithm 1 step 2. For convenience, we will refer to these as DNA alignments and RNA alignments, respectively. High-quality alignments were ensured by retaining only reads with mapping quality scores
Gene prediction and ortholog assignment
Following de novo assembly, protein-coding genes were identified and taxonomically annotated with eggNOG-mapper v2.2 (database 5.0) in metagenome mode (Cantalapiedra et al., 2021). Step 3 in Algorithm 1 yields, for every sample, (i) a General Feature Format (
RNA editing detection and DNA mutations filtering
In ME
Mapping edits to genes and orthologs
Because each metagenomic assembly is sample-specific, contig identifiers, and hence the genes predicted on those contigs, are unique to a single library. To place RNAediting events into a common coordinate system, in step 5, we first converted every edit position from contig space to gene space: the GFF file for each sample was parsed to obtain the start, end, and strand of every coding sequence, the raw contig coordinate of the edit was verified to fall within a Coding Sequence, and its 1-based offset from the gene’s 5-prime end was calculated with strand correction when required. The resulting intragenic coordinates were then grouped by eggNOG seed ortholog, and the corresponding protein sequences were aligned with MAFFT (Katoh and Standley, 2013), allowing orthologous editing sites from different samples to be projected onto the same column of the multiple-sequence alignment for downstream comparative analyses.
Edit-site filtering and cross-sample validation
To ensure the robustness of identifying RNA editing events, in step 6, we implemented a multi-tiered filtering and validation framework. A candidate edit site was retained only if it was observed in at least five independent samples, each meeting stringent read support criteria: a minimum of
To confirm positional homology across genomes, we leveraged multiple sequence alignments generated using
The resulting high-confidence set of candidate edits was subjected to statistical evaluation (step 7) to distinguish true biological modifications from sequencing artifacts or stochastic variation. This rigorous filtering process produced a conservative and reliable catalog of reproducible RNA editing events across the microbiome, suitable for comparative and functional analyses.
Functional annotation and impact prediction
To elucidate the biological relevance of the curated RNA editing catalog, we combined the seed-ortholog assignments (see Section 2.4) with domain, pathway, and taxonomic metadata provided by eggNOG-mapper(Cantalapiedra et al., 2021). Each edited ORF was linked to its KEGG ortholog, COG category, and taxonomic lineage, enabling pathway-level and clade-specific summaries of editing frequency.
Protein consequences were evaluated in two steps. First, both the reference and edited coding sequences were translated in silico with BioPython (Cock et al., 2009) under the bacterial genetic code. Second, each codon change was classified as synonymous, missense, or nonsense; missense substitutions that overlapped residues annotated by eggNOG-mapper as catalytic or ligand-binding sites were flagged as potentially disruptive (Cantalapiedra et al., 2021).
Together, this functional annotation framework provides a comprehensive understanding of the biological context and potential consequences of RNA editing in the microbiome—spanning gene function, evolutionary conservation, taxonomic origin, pathway participation, and protein-level impact.
Statistical analysis
To assess the statistical significance of RNA edit sites, we employed a multi-layered approach integrating both site-level and gene-level analyses. For each candidate RNA editing site, we applied a binomial test to evaluate whether the proportion of edited bases observed in RNA reads significantly exceeded what would be expected by random sequencing error alone. The null hypothesis assumes that the observed editing proportion is due to background noise, while the alternative supports a true post-transcriptional modification.
In addition to evaluating individual sites, we conducted differential RNA editing analyses across disease and control groups to identify editing sites associated with clinical phenotypes. This analysis was performed at two levels: (i) site-level differential editing, comparing RNA editing scores across cohorts using non-parametric Kruskal–Wallis tests, and (ii) gene-level aggregation, where editing scores from multiple sites within the same gene were combined to assess broader patterns of regulation. This gene-centric view accounts for the potential clustering of edits within functionally important regions and reduces site-level noise.
To adjust for the large number of statistical tests performed across the transcriptome, we implemented the Benjamini–Hochberg procedure (Benjamini and Hochberg, 1995) to control the FDR. Sites or genes with FDR-adjusted p values below 0.1 were considered statistically significant. Furthermore, we performed taxon-specific RNA editing analyses to investigate whether certain bacterial clades—defined at the family or class level—exhibited unique editing profiles. This stratified analysis enabled the identification of clade-restricted RNA editing signatures, offering insights into potential lineage-specific post-transcriptional regulatory mechanisms within the microbiome. Different false discovery FDR thresholds were applied depending on the level of analysis: site-level tests used FDR below 0.05, whereas gene-level and other aggregated analyses used FDR below 0.1, reflecting the reduced multiple-testing burden and the exploratory aim of integrative analyses.
Code availability and tool versions
The ME
RESULTS
Starting from a dataset of 748 paired metagenomic and metatranscriptomic samples collected from 96 individuals, ME
High-Confidence Missense RNA Edit Sites in Genes Previously Reported to Undergo Editing
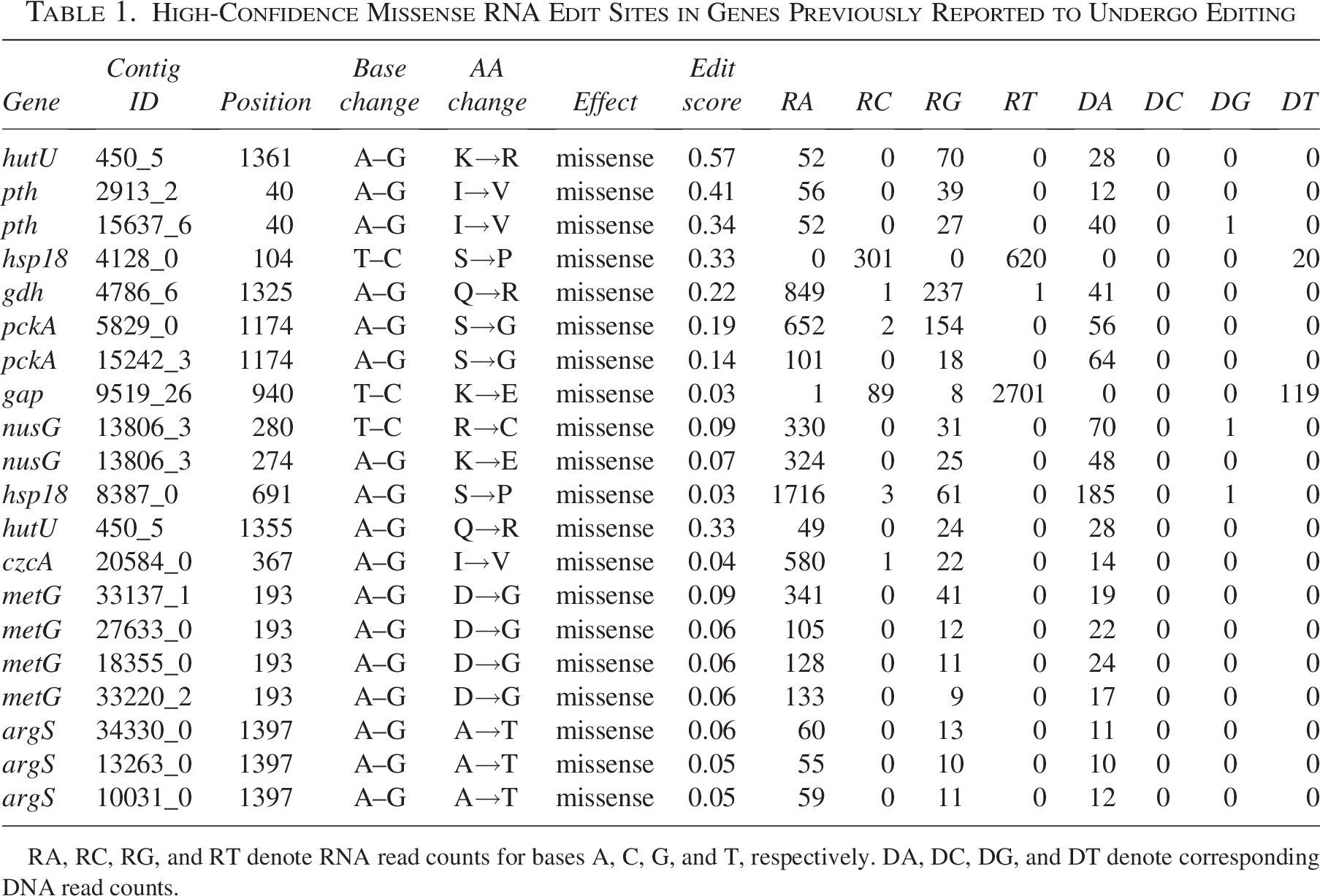
High-Confidence Missense RNA Edit Sites in Genes Previously Reported to Undergo Editing
RA, RC, RG, and RT denote RNA read counts for bases A, C, G, and T, respectively. DA, DC, DG, and DT denote corresponding DNA read counts.
The distribution of the 2,295 RNA edit sites across the microbiome was highly family-specific, with 11 bacterial families accounting for the majority of edit sites (see Fig. 2). The top 7 families consisting of Bacteroidaceae, Ruminococcaceae, Rikenellaceae, Porphyromonadaceae, Lachnospiraceae, Clostridiaceae, Eubacteriaceae, had over 2100 of the 2,295 edit sites. Three out of the seven families mentioned above are from the order Bacteroidales in the phylum Bacteroidota, making the order Bacteroidales represent the majority (1711) of the edit sites. The other four are from the class Clostridia in the phylum Bacillota. This non-uniform distribution suggests that RNA editing is not a random occurrence but may represent a lineage-specific regulatory mechanism active in select microbial groups, with particular emphasis on the order Bacteroidales and the class Clostridia. A similar distribution of the count of edit sites by phylum and class is shown in the Appendix (see Figs. A1 and A2). It is worth noting that the edit sites are most abundant in the phylum Bacteriodota with a distant second in the Firmicutes. A minuscule number were found in the phylum Proteobacteria.
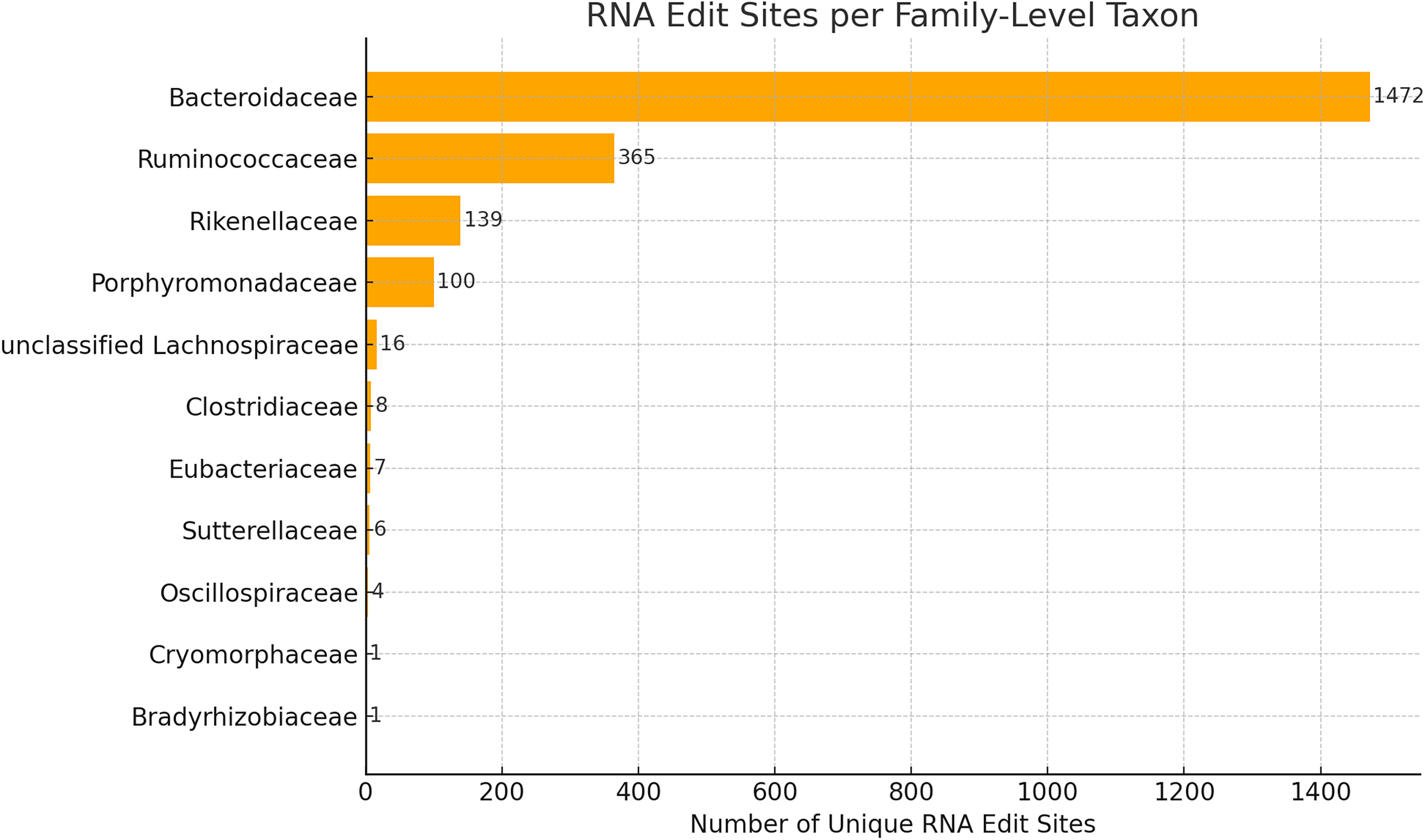
Family-level distribution of RNA editing sites across the gut microbiome. Bar plot showing the number of unique high-confidence RNA editing sites assigned to bacterial families. Editing sites are non-uniformly distributed, with Bacteroidaceae and Ruminococcaceae contributing the majority of events, consistent with their high abundance in the Integrative Human Microbiome Project (iHMP2) samples.
Importantly, the top four families were also among the most abundant taxa in the iHMP2 dataset, as reported in previous analyses of gut metagenomes from individuals with and without IBD (Lloyd-Price et al., 2019). For example, Bacteroidaceae alone accounted for over 65% of average microbial abundance in healthy individuals, while Ruminococcaceae and Lachnospiraceae were consistently detected at moderate-to-high abundance across all cohorts. The co-occurrence of high abundance and elevated RNA editing activity in these taxa strongly suggests that editing may play a functional role in modulating gene expression or adaptation in dominant gut microbes.
The edit site number distributions at other levels of the taxonomic hierarchy are provided in the Appendix.
To investigate whether individual RNA edit sites differ between disease states, we performed site-level differential analysis using the Kruskal–Wallis test across three diagnostic groups: UC, CD, and controls (non-IBD). After adjusting for multiple comparisons using the Benjamini-Hochberg method, no individual RNA edit sites exhibited statistically significant differences (FDR
RNA editing in highly conserved genes across multiple samples
To explore whether RNA editing patterns are functionally meaningful at broader biological scales, we aggregated RNA editing sites at the gene level, enabling a more integrated analysis of editing activity within entire coding regions. While this analysis ignores the microbial taxa in which the editing may be occurring, such an analysis is in line with the broader hypothesis that microbiomes act in a coordinated fashion where an entire community may be holding receptacles of gene families and consequently imparting functional capabilities to the collective. This approach accounts for the tendency of RNA editing in bacteria to occur in clusters within genes (Bar-Yaacov et al., 2017; Nie et al., 2020), rather than as isolated single-nucleotide changes. Clustered editing also allows for the resulting proteins to have more changes than what may be possible with a single nucleotide edit.
We investigated whether RNA editing preferentially targets functionally important genes by focusing on identifying highly conserved bacterial genes that exhibit multiple RNA editing sites across a set of microbiome samples from multiple samples. This analysis aimed to determine whether essential microbial functions, typically encoded by evolutionarily stable genes, are subject to post-transcriptional modifications. Our analysis identified several genes exhibiting extensive RNA editing across numerous samples. Notably, genes such as dnaK (Heat shock 70 kDa protein), gap (Glyceraldehyde-3-phosphate dehydrogenase), groL (Chaperonin), and pckA (Phosphoenolpyruvate carboxykinase) demonstrated both a high number of RNA edit sites and widespread occurrence across more than 200 samples each, indicating pervasive RNA editing activity across diverse microbial populations. The genes with the highest number of edit sites in a large number of samples are shown in Figure 3.
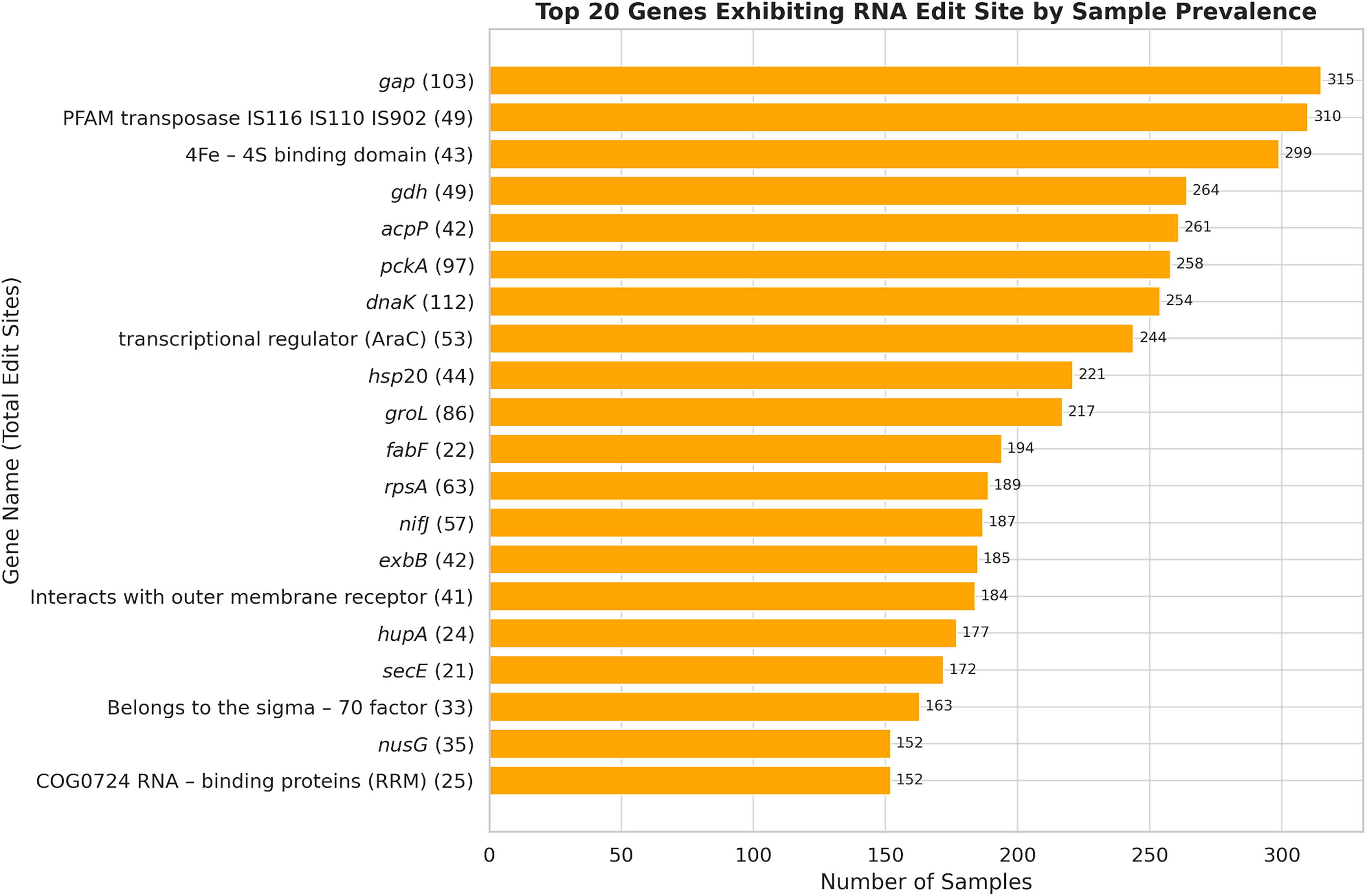
Highly conserved genes exhibit widespread RNA editing across samples. Top 20 genes ranked by the number of samples in which RNA editing was detected. Bars indicate the number of samples containing an editing site within that gene, with total edit-site counts shown in parentheses. Many of these genes (e.g., dnaK, gap, groL, pckA) encode essential metabolic or stress-response functions.
Among these highly edited genes, several encode universally conserved microbial proteins, including dnaK, groL, and gap. These genes perform essential cellular functions such as protein folding, stress responses, glycolysis, and energy metabolism, and their evolutionary conservation is well established in bacterial phylogenetic studies (Woese et al., 1990; Ciccarelli et al., 2006; Jordan et al., 2002). The pronounced RNA editing observed in these conserved genes points to an additional regulatory complexity layer previously underappreciated in microbiological research.
Conventionally, highly conserved genes maintain low sequence variability at the DNA level to preserve essential cellular functions (Jordan et al., 2002). However, our findings reveal frequent RNA edit sites in these conserved genes, suggesting RNA editing as a potential adaptive mechanism allowing transient functional diversification without permanent genomic alterations. Previous studies have highlighted role of RNA editing in microbial adaptation, particularly under environmental stress conditions (Bar-Yaacov et al., 2017; Nie et al., 2020).
The widespread and consistent RNA editing observed in metabolic enzymes, notably gap and pckA, aligns with previous reports identifying these enzymes as critical under nutrient-limiting and inflammatory conditions frequently associated with diseases such as IBD (Shepherd et al., 2018).
Collectively, our data strongly suggest that RNA editing preferentially targets conserved bacterial genes involved in vital cellular processes. The frequent and widespread editing across multiple taxa and samples underscores the role of RNA editing as a dynamic regulatory strategy, potentially facilitating rapid microbial adaptation to environmental challenges or host-associated conditions.
Next, we investigate the relationship between clusters of edit sites and disease. Using mean RNA edit scores across the genes normalized by approximate gene length, we performed differential RNA editing analysis at the gene level across the three diagnostic groups: UC, CD, and non-IBD controls. After correcting for multiple hypothesis testing using the Benjamini–Hochberg procedure, several genes exhibited significant differences in editing levels at FDR < 0.1. Figure 4 presents a heatmap of the mean gene-level RNA editing scores in the three cohorts. Among the genes showing differential editing, bcd2 (acyl-CoA dehydrogenase, involved in fatty-acid
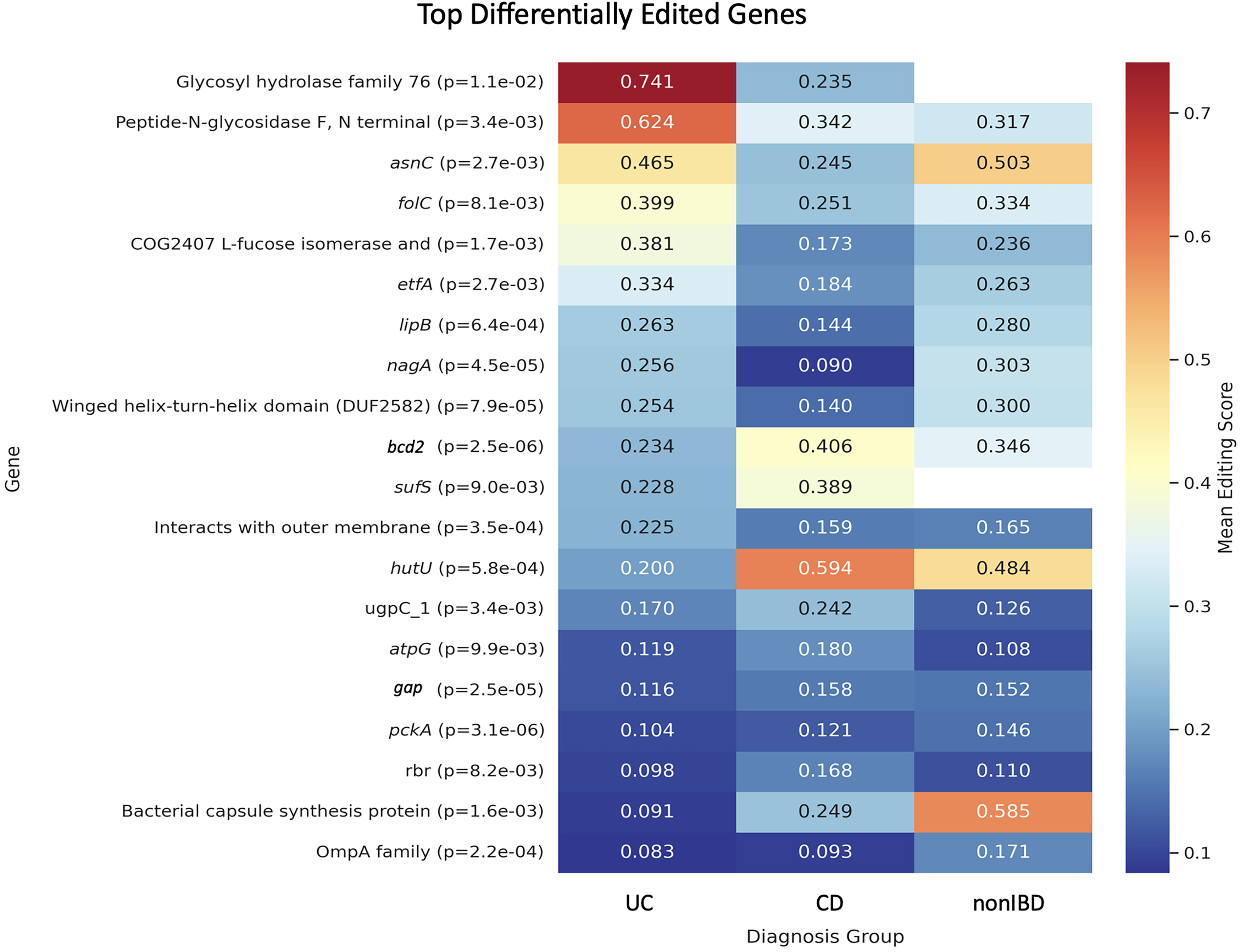
Heatmap of mean gene-level RNA editing scores for selected biologically relevant genes across ulcerative colitis (UC), Crohn’s disease (CD), and non-inflammatory bowel disease (nonIBD) controls. Editing scores represent the mean aggregated site-level editing normalized by gene length. Genes shown passed false discovery rate (FDR) < 0.1 using Kruskal–Wallis testing. Color scale indicates relative editing intensity.
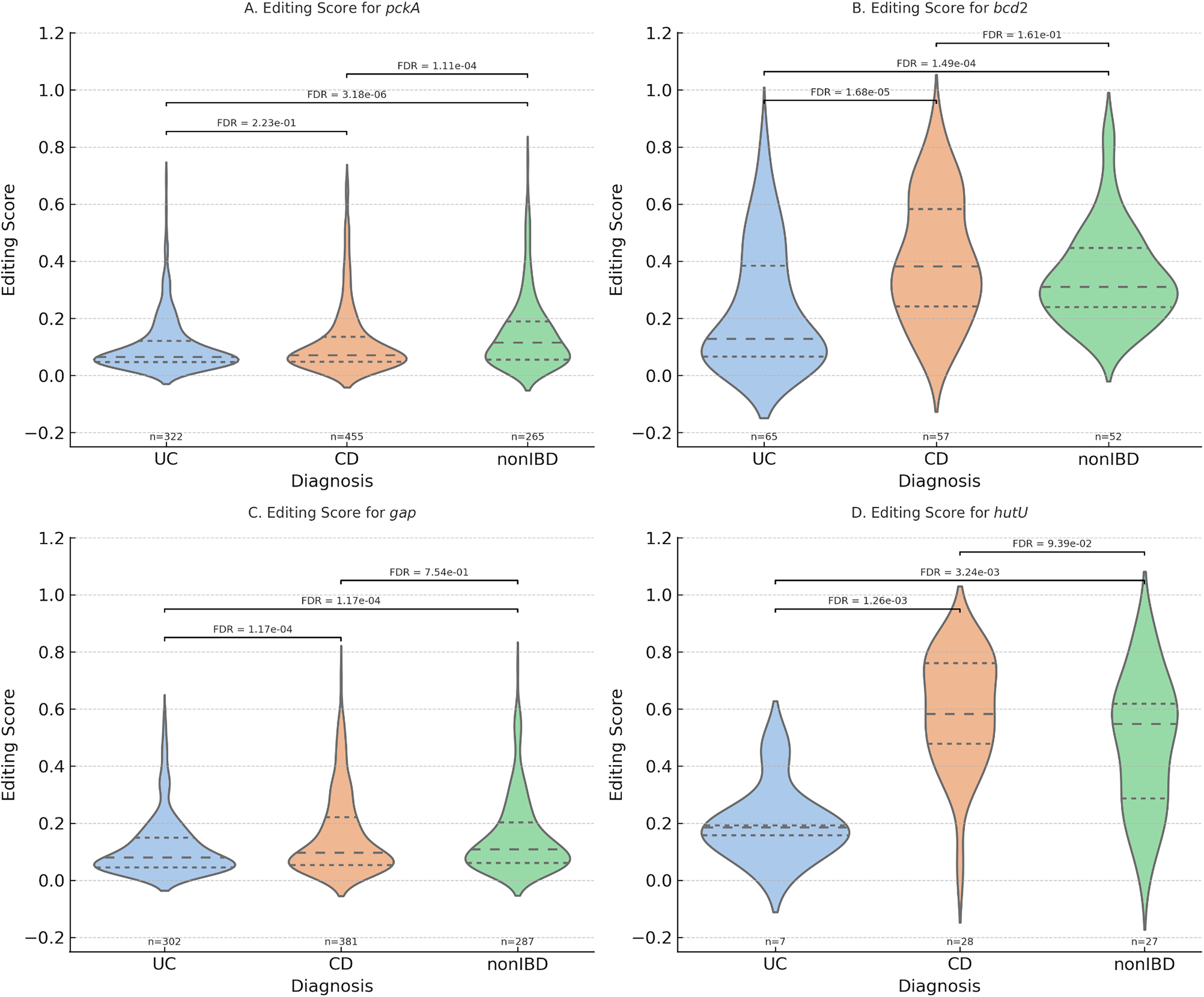
Disease-associated differences in RNA editing for key metabolic genes. Violin plots showing the distribution of gene-level RNA editing scores for pckA, bcd2, gap, and hutU across UC, CD, and non-IBD cohorts. Each point represents an individual sample. Group differences were assessed using pairwise Mann–Whitney U tests with FDR correction; adjusted p values are shown.
These results support the hypothesis that RNA editing may be involved in adaptive transcriptional regulation of bacterial gene function in response to host-associated conditions. The disease-specific editing patterns observed in metabolic genes like gap and pckA align with previous reports describing metabolic shifts and microbial functional reprogramming in the gut microbiome during IBD progression (Lloyd-Price et al., 2019). Collectively, our gene-level analysis suggests that RNA editing contributes to functional modulation of microbial activity, potentially enhancing the ability of gut bacteria to respond dynamically to inflammatory environments.
Further stratifying the analysis by taxonomic groups highlighted distinct differential editing patterns at the gene level in each family. Within the Ruminococcaceae family (see Fig. 6) the gene nagA (N-acetylglucosamine-6-phosphate deacetylase, involved in amino sugar metabolism) exhibited significantly lower editing levels in CD samples, while the Bacteroidaceae family (see Fig. 7) showed increased hsp20(small heat shock protein) editing in UC samples. These taxon-specific patterns illustrate the nuanced interplay between bacterial taxa and host disease states, suggesting that RNA editing within specific taxa contributes to functional adaptations of the microbiome, potentially influencing the progression and severity of IBD.
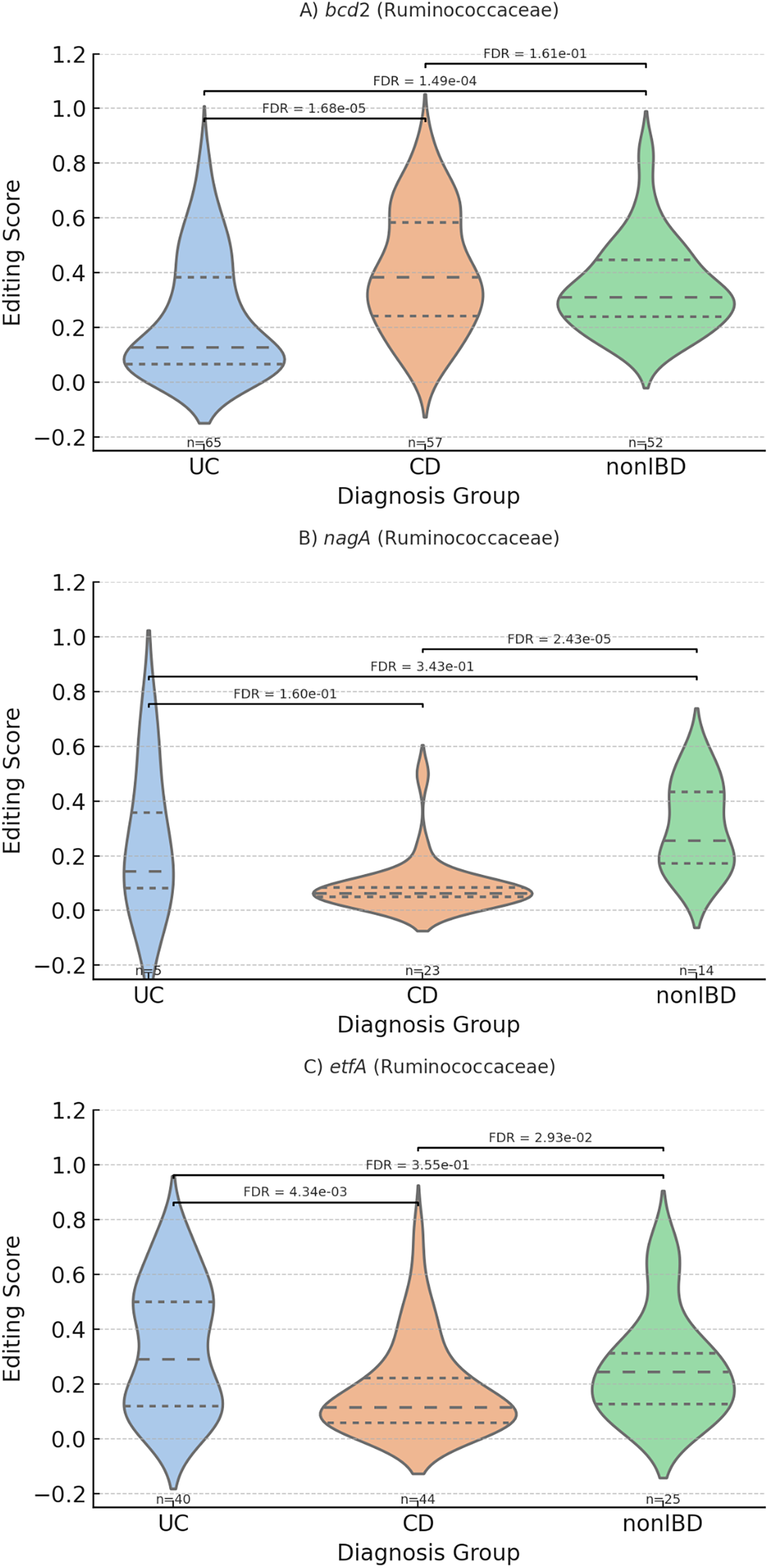
Taxon-specific gene-level RNA editing patterns. Violin plots showing RNA editing scores for selected genes within the Ruminococcaceae family across diagnostic groups. These analyses highlight lineage-specific RNA editing differences that are masked in global gene-level aggregation. Statistical testing was performed using Mann–Whitney U tests with FDR correction. Adjusted p values are shown.

Taxon-specific gene-level RNA editing patterns. Violin plots showing RNA editing scores for selected genes within the Bacteroidaceae family across diagnostic groups. These analyses highlight lineage-specific RNA editing differences that are masked in global gene-level aggregation. Statistical testing was performed using Mann–Whitney U tests with FDR correction. Adjusted p values are shown.
Additional violin plots for significantly differentially edited genes from other families (including Porphyromonadaceae) are shown in Appendix Figure A3.
Our study highlights RNA editing as an important mechanism for microbial adaptation within the human gut microbiome. Utilizing a de novo assembly approach allowed unbiased detection of editing sites across diverse, understudied bacterial taxa and unclassified novel strains. Enrichment of RNA editing in family-level taxa like Bacteroidaceae and Ruminococcaceae suggests adaptive responses to inflammatory conditions, supporting prior associations with functional dysbiosis in IBD.
We validated our predictions in two ways: (i) overlap with previously confirmed edits in E. coli (Bar-Yaacov et al., 2017; Mehta et al., 2025), and (ii) stringent multi-sample filtering that excludes DNA mutations. Nevertheless, experimental validation (e.g., Sanger sequencing, strand-specific RNA-seq) will be critical in future studies. A limitation of our approach is that low-abundance taxa with fragmented assemblies may be underrepresented due to coverage filters. While this reduces false positives, it may underestimate editing diversity in rare microbes. Also, our statistical models do not currently adjust for host metadata (e.g., age, medication, sequencing depth), as these data points were sparse in iHMP2. Future work with linear mixed models will better account for confounders in experiments designed to study RNA editing.
The observed taxon-specific RNA editing indicates evolutionary adaptations tailored to distinct bacterial metabolic and physiological demands, aligning with previous isolated bacterial studies. Despite extensive analyses, we did not identify significant differential editing at individual sites across disease states, suggesting subtle site-level variations or broader cluster-based effects, consistent with the clustered RNA editing patterns described in the literature.
Notably, significant gene-level editing in stress-response and metabolic genes, including dnaK, gap, pckA, and groL, highlights their potential roles in bacterial adaptation to inflammatory stress. To further contextualize these findings, we report effect sizes for gene-level differential RNA editing as both absolute differences in mean editing scores and log2 fold changes (Supplementary Table S1). Genes exhibiting the largest effect sizes encode enzymes central to microbial metabolism and stress adaptation. For example, gap (glyceraldehyde-3-phosphate dehydrogenase), a key glycolytic enzyme involved in energy production, showed reduced RNA editing in ulcerative colitis relative to non-IBD controls (
In conclusion, our findings underscore RNA editing as a critical, dynamic, and taxon-specific regulatory mechanism in the gut microbiome, warranting future experimental validation and functional studies to elucidate its therapeutic potential.
AUTHORS’ CONTRIBUTIONS
A.M.: Conceptualization, methodology, software, formal analysis, investigation, data curation, visualization, writing—original draft. V.S.: Methodology, software, formal analysis, writing—review and editing. K.M.: Resources, biological interpretation. G.N.: Conceptualization, supervision, project administration, funding acquisition, writing—review and editing.
Footnotes
ACKNOWLEDGMENT
The authors thank ICCABS 2025 for the invitation to write for the JCB (Journal of Computational Biology, SI. The authors also thank the Integrative Human Microbiome Project (iHMP2) consortium for generating and making available the multi-omics datasets used in this study. The authors also acknowledge the Florida International University High Performance Computing resources for computational support.
AUTHOR DISCLOSURE STATEMENT
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
FUNDING INFORMATION
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Supplemental Material
Appendix
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.