
Abstract
The prediction of protein–protein interaction (PPI) can be insightful for exploring the molecular mechanisms of cellular functions. Constructing the negative datasets of PPI is related to the assessment of the prediction accuracy and evaluation of the prediction performance. Aiming at the problem of unstable prediction accuracy in the current method of building negative sets using random sampling, we proposed a method of constructing negative sets based on a conditional generative adversarial network (CGAN), named PPIGAN. This method generates negative samples through a generative network, and the PPI prediction model uses these generated negative samples along with positive samples to learn interaction features. Simultaneously, the generator and the prediction model continuously compete against each other during the learning process, which enhances the model’s generalization ability and prediction accuracy. Experimental results show that the accuracy of our proposed method reaches 94.68% and 98.22% in 5-fold cross-validation on yeast and human datasets, respectively. These results either surpass or closely approach the performance of advanced PPI prediction models such as PIPR, convolutional neural network, DeepTrio, and DeepFE, indicating that the method proposed in this article provides an effective solution for the work related to PPI prediction.
Keywords
INTRODUCTION
Protein–protein interactions (PPIs) can reveal the roles of organisms at the molecular level and help to reveal the regulatory mechanisms of biological growth and development, environmental stress, signal transduction, and so on (Hosseini et al., 2024; Lin et al., 2023). To address the challenge of producing a large number of PPIs, extensive research has been conducted (Elizabeth et al., 2023; Zhang et al., 2023). Some high-throughput experimental methods have been proposed, such as yeast-two-hybrid, bimolecular fluorescence complementation, biochemical modification analysis and cocrystal structure analysis (Lage, 2014; Oughtred et al., 2019). Although these experimental methods have high detection accuracy, they are limited by the scale of proteins detected in each experiment and are time-consuming and labor-intensive (Hu et al., 2024). Therefore, researchers have attempted to develop computational PPI prediction methods to reveal molecular mechanisms, thereby greatly reducing the required time and costs (Song et al., 2022; Szymborski et al., 2022; Wimalagunasekara et al., 2023).
With the rapid development of proteomics, the protein sequences of many organisms have been determined. Some computational methods for predicting PPIs based on protein sequences have been developed (Chen et al., 2019; Hu et al., 2022). Deep learning models, consisting of multiple linear or nonlinear models, simulate advanced abstractions through deep mappings involving multiple layers, and obtain good performance. The greatest advantage of deep learning over traditional machine learning approaches is its ability to automatically extract abstract and hard-to-capture features, which is beneficial for deep mining of the intrinsic information of sequences and improving prediction performance (Liu et al., 2023).
Based on deep learning methods, Sun et al. (2017) proposed the use of a stacked autoencoder algorithm as a classifier for PPI, building on the classification of amino acids from Shen et al. (2007) and Guo et al. (2008). This algorithm comprises a deep neural network (DNN) model with multiple layers of sparse autoencoders, and the prediction results indicate that deep learning methods outperform traditional machine learning methods such as random forests and support vector machines in both model performance and training time. The DPPI (Hashemifar et al., 2018) architecture based on convolutional neural network (CNN) extracts protein’s local features from protein configuration files, and this architecture demonstrates superior predictive performance within and across species. The DNN–PPI (Li et al., 2018) with two independent CNN encoders extracts features from amino acid sequences using CNN and long short-term memory (LSTM) networks . Chen et al. (2019) proposed an end-to-end framework called PIPR (Protein-Protein Interaction Prediction Based on Siamese Residual RCNN), which only utilizes protein sequences for protein interaction prediction. PIPR uses pretrained vectors to represent amino acids, capturing their contextual similarity and physicochemical properties, and adopts a residual recurrent CNN (RCNN) as the model architecture. In addition to predicting PPI, PIPR also proposes interaction type prediction and affinity estimation models for different application scenarios. Hu introduced a sequence-based method called DeepTrio for PPI prediction (Hu et al., 2022); this method employs multiple parallel CNNs to comprehensively utilize protein sequence features, achieving a prediction accuracy of 98% on human protein-protein interactions. Chen proposed a novel neural network called TransformerCPI (Chen et al., 2020), which demonstrated excellent performance in deconvolution to highlight important interaction regions between protein sequences and compound atoms. This method adopts a transformer structure, effectively capturing interactions between one sequence and another through self-attention mechanisms, and provides new insights for sequence feature extraction.
Although existing methods achieve good performance in PPI prediction, there is still no “gold standard” available for the negative dataset in PPI prediction. Negative dataset is crucial for assessing the reliability of prediction models and objectively evaluating prediction accuracy. It is challenging to obtain noninteracting protein datasets through biological experimental methods because it requires extensive experiments to sufficiently validate the inertness of proteins. Despite existing methods for constructing negative datasets such as “general random sampling”(Pan et al., 2010), “balanced random sampling”(Yu et al., 2010), “subcellular localization”(Shen et al., 2007), and “shuffle protein sequences”(Hu et al., 2022), there is still room for further improvement in prediction accuracy.
Generative adversarial networks (GANs) are proposed as a computational model that can generate pseudo-samples as close as possible to real samples, improving the discriminator’s ability to distinguish between real and fake samples(Goodfellow et al., 2020). By introducing noise into deep learning models based on the GAN concept, high-quality negative sample data can be generated, effectively enhancing feature learning models and feature representation capabilities(Arjovsky et al., 2017; Wang et al., 2023). Wan and Jones (2020) successfully utilized GANs to generate biophysical properties from protein sequences. Meanwhile, Repecka et al. (2021) established ProteinGAN based on an attention mechanism, which learns evolutionary relationships of protein sequences from complex multidimensional amino acid sequence spaces, creating new, diverse sequences with natural physical properties. Li proposed a GAN-based method for predicting phage–host interactions (PHI) (Li and Zhang, 2022), which employs a GAN-based data augmentation module to generate fake PHIs to increace richness of data.
In the current study, we proposed a model called PPI based on GAN (PPIGAN) for constructing negative sets based on a GAN for downstream sequence-based PPI prediction tasks. PPIGAN employs a generator, which introduces a protein from the positive samples as a condition to generate a fake protein sequence to construct negative samples, in order to generate a more sensitive and discriminative prediction model and improve the model’s generalization ability and robustness. Experimental results show that the negative sample generation method proposed in this study achieves competitive overall PPI prediction performance and yields significant performance improvements over existing mainstream PPI methods on real negative benchmark datasets. PPIGAN is available at: https://github.com/jiantaoyuNWAFU/PPIGAN.
MATERIALS AND METHODS
Datasets
The choice of dataset plays a crucial role in model construction, the Saccharomyces cerevisiae (yeast) and the Homo sapiens (human) data are widely used to evaluate the performance of PPI predictors (Chen et al., 2019; Hu et al., 2022; Yao et al., 2019). In addition to these two benchmark data sets, we added the high-quality BioGRID human dataset proposed by Hu et al. (2022) and the interactions between viruses and human proteins proposed by Wang et al. (2021) as the test dataset to comprehensively evaluate the generalization ability of the model (as shown in Table 1).
The yeast dataset, as a widely used benchmark, is composed of 5594 positive PPI samples proposed by (Guo et al., 2008). The positives are selected from the database of interacting proteins, DIP_20070219. The protein sequences are retrieved from the UniProt (Consortium, 2019), where proteins shorter than 50 amino acids are removed. And CD-HIT (Cluster Database at High Identity with Tolerance) is used to cluster according to the clustering criterion of homogeneity The human dataset, sourced from HPRD database, is composed of 36,630 unique positive samples, by eliminating self-interactions and repetitive interactions (Pan et al., 2010). Similar to yeast sequence processing, 10,370 human proteins are retained. The BioGRID–human dataset. This dataset is a curated human interaction dataset derived from the BioGRID database. It consists of 31,164 pairs of samples involving 7705 proteins. All protein sequences are obtained from UniProt, and the length of the sequence is limited to 150–1500 amino acids. In order to reduce the sequence redundancy, consistent with the processing method of the benchmark datasets, CD-HIT is used to process the dataset, and 40% sequence similarity is set as the clustering threshold. The virus–human test set. Similar to the method used in DeepTrio, the virus and human protein interaction dataset proposed by Wang et al. (2021) is also used as an independent test set in this article to evaluate the performance of PPIGAN and other methods. To reduce redundancy, viral sequences in the independent test set showing >25% sequence similarity to the BioGRID human training set were excluded. The final independent test set consists of 17,858 pairs of proteins involving 4790 proteins.
Collected Positive Datasets

PPI, protein–protein interactions
GANs, as a kind of deep learning model, is one of the most promising methods in unsupervised learning on complex distributions in recent years (Goodfellow et al., 2020). They aim to learn the joint probability distribution of training data, capturing underlying information within the data distribution space (Xu et al., 2020). Consequently, GANs can generate new samples with features resembling those of the training data, widely applied in the field of bioinformatics (Hasegawa et al., 2022). Researchers improved the training algorithm and the network structure of GAN, and a large number of GAN variants have emerged (Huang et al., 2023; Rather and Kumar, 2023; Wang et al., 2022). The conditional GAN (CGAN; Yang, 2020) is one of these variants. CGAN transform unsupervised original GANs into supervised learning by introducing a conditional variable to restrict GAN generation. Conditional variable is added to any layer of the generator and discriminator as an additional input to guide and constrain the sample generations. The network will learn to adapt to these additional inputs by adjusting its parameters.
Figure 1 illustrates the flowchart of PPIGAN. It is a supervised deep neural learning network with set conditions, which includes a generative network and a discriminative network. For a pair of interacting proteins

The flowchart of PPIGAN. For a pair of interacting proteins, where one protein is selected with higher degrees (more interactions in the PPI network), the generator takes it along with the noise as input and generates a protein sequence embedding vector. The selected protein and the generated protein form a negative sample, which is fed into the PPI prediction module together with the positive sample to learn protein interaction features, and output binary classification probabilities.
The method of building negative sets has always been an important issue, which is related to the assessment of prediction accuracy and the evaluation of the performance of prediction methods (Yu et al., 2023). However, it is difficult to obtain noninteracting protein datasets through biological experimental methods, because researchers need to do a large number of biological experiments to verify the inertness between proteins. The most commonly used method for building negative sets is the random sampling approach. The idea of this method is that, given the pairs of interacting proteins, two proteins are randomly selected from the remaining protein pairs to form a negative sample. However, the predictive performance of this method is not always stable (Lannelongue and Inouye, 2024).
In order to generate negative samples, we used the encoder–decoder structure in the generative network, which transforms the protein sequences into dense low-dimensional vectors, and fuses them with the noise z as the input of the generative network. The structure of the generative network is shown in Figure 2.
A diagram of the generator’s structure. The protein sequence is first converted into a vector and then concatenated with noise z as input to the generator. A 10-layer convolutional neural network is used to learn the sequence features, and a 10-layer deconvolutional neural network is used to generate a new protein sequence.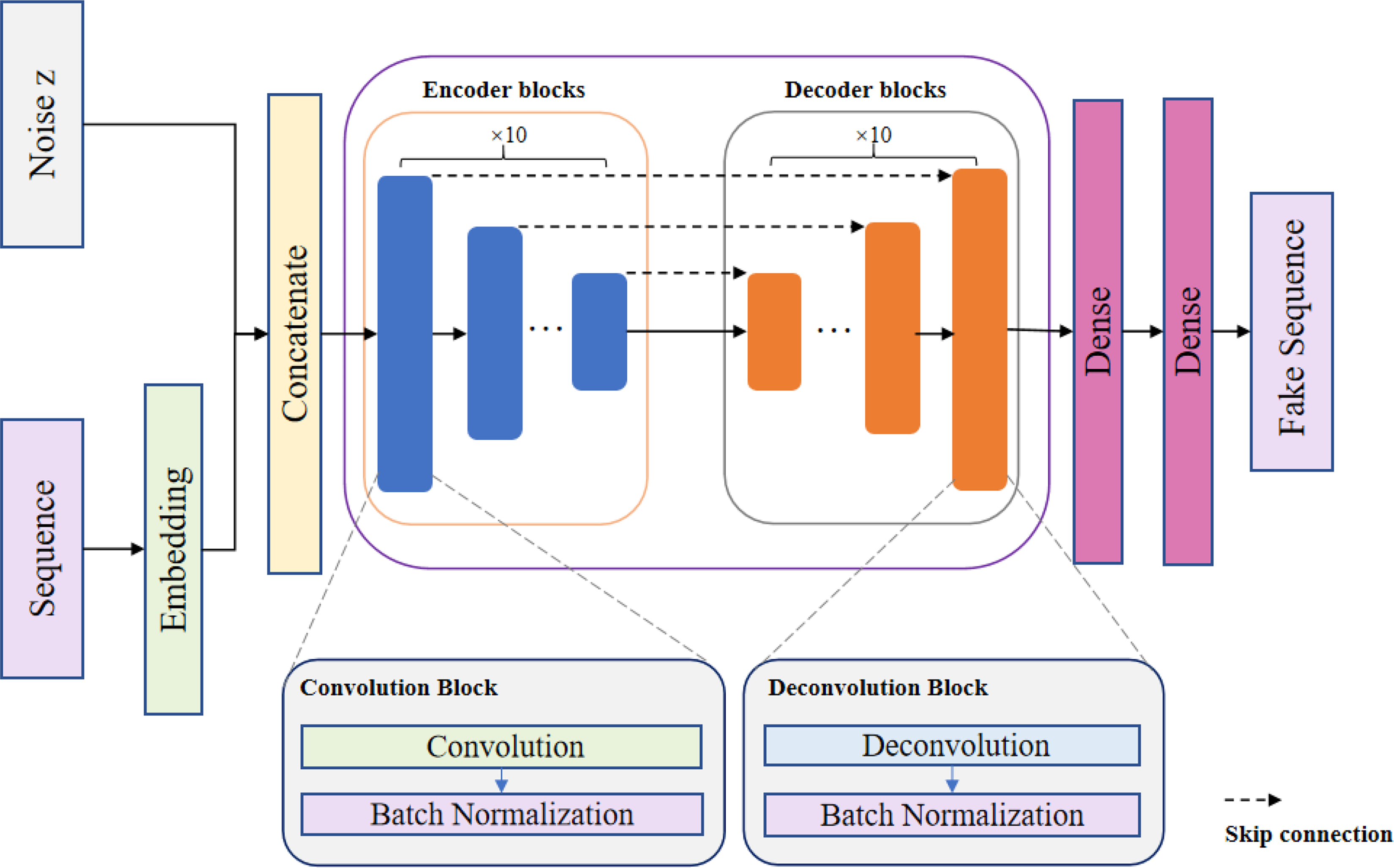
The generator consists of an encoder and a decoder. The encoder uses a 10-layer convolutional structure to gradually reduce the spatial dimension of the input protein embedding vector and learn its distribution characteristics. The decoder gradually restores the details and spatial dimensions of the protein embedding vector through a 10-layer deconvolution structure. The generator converts the random noise of the input network into a new protein sequence under the condition of protein sequences. The goal of the generative network is to generate a new protein sequence that will interact with the input protein sequence. This network adopts fully-connected skip connections, with direct connections between all input and output layers. This connection method not only fuses low-level and high-level features but also accelerates information propagation through the network, which helps alleviate the problem of gradient vanishing. The specific steps are as follows.
Encoding block: 10 layers of convolution are used for down-sampling, and a batch normalization layer (Ioffe and Szegedy, 2015) is introduced in order to alleviate the gradient vanishing and accelerate the network convergence. Each convolutional structure contains one convolutional layer as well as a batch normalization layer with a LeakyReLU activation function, where the length of the convolution kernel is 3. The number of channels is reduced by half after each down-sampling to obtain the coded features. Decoding block: 10 layers of deconvolution are used for up-sampling the coded features obtained in (1). Each layer in the deconvolutional structure is symmetrically connected to each layer in the convolutional structure, thus completing the decoding process. With skip connections, the loss of protein sequence features in the decoding process can be avoided, which increases the credibility of the model. The LeakyReLU activation function expression is as follows.
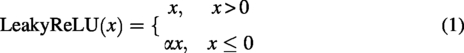
Where
To describe protein sequences, the set A is used to represent 20 amino acids. A protein can be represented as an amino acid sequence
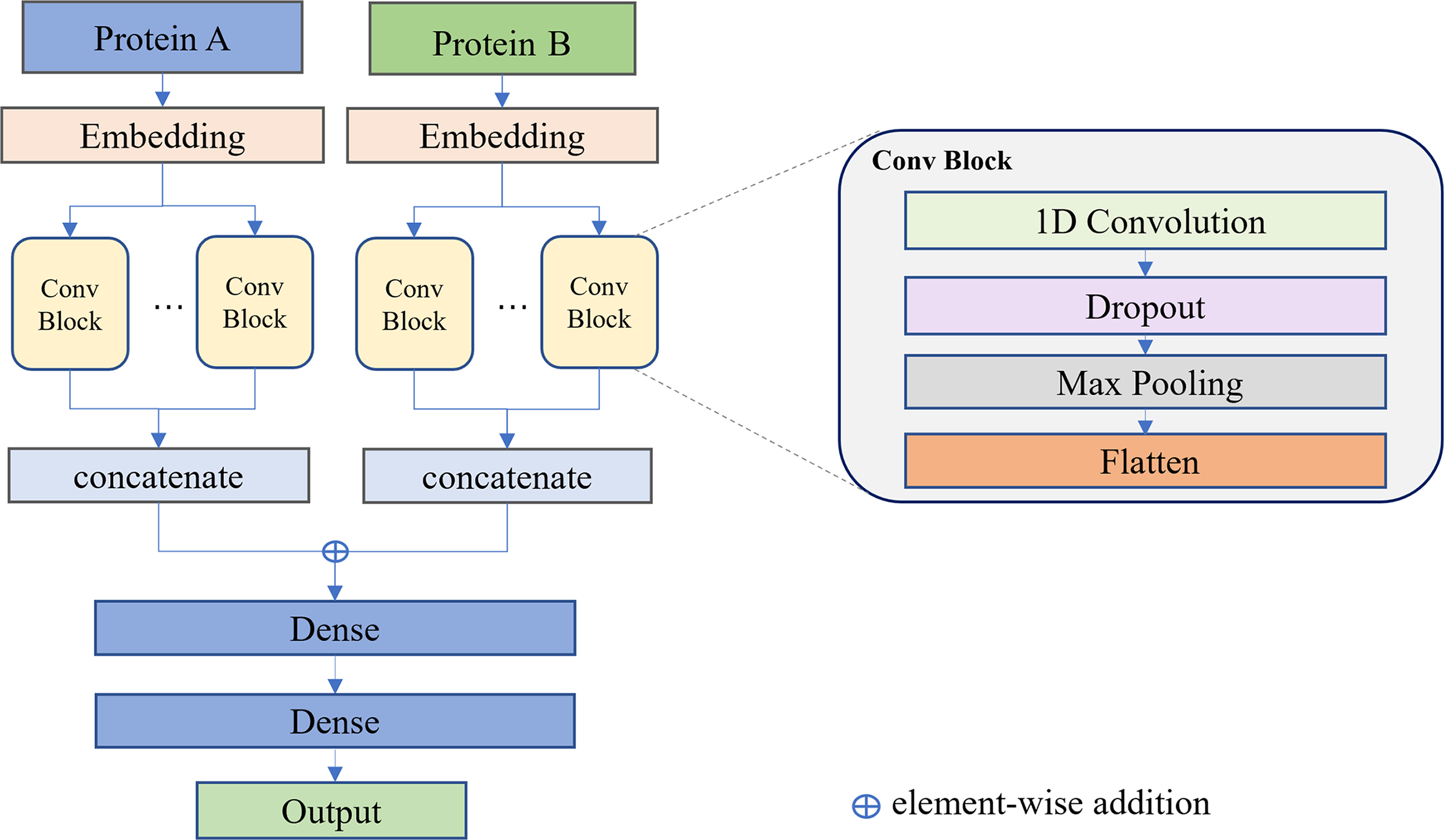
A diagram of the discriminator’s structure. The protein sequences A and B are inputted and converted into embedding vectors. These vectors are then fed into multiple parallel convolution modules with different kernel sizes to extract features. The multi-scale features are flattened and concatenated. The feature vectors of two proteins are element-wise added and then pass through two dense layers for classification.
In order to process sequences, it is necessary to convert them into embedding vectors. In this article, each amino acid is transformed into a d-dimensional vector, resulting in a 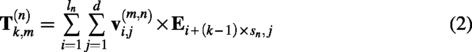

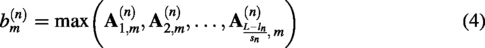
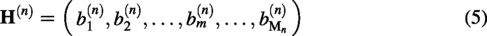
All experiments were conducted using the PyTorch 1.12 framework, with coding done in Python 3.7. The hardware environment for the experiments consisted of a single NVIDIA GeForce RTX 3080Ti GPU with 12GB RAM. The model parameters were optimized using the Adam optimizer, with the learning rates for both the generator and the discriminator set to 0.0001. Additionally, both the generator and discriminator employed the cross-entropy loss function. The model was trained for 100 epochs using a 2:1 update ratio between the discriminator and generator.
Performance evaluation
To validate the generalization ability of the model, a common metric is needed for assessment. We employ several commonly used evaluation measures to assess the performance of PPIGAN and other sequence-based state-of-the-art methods, including accuracy, specificity, sensitivity, F1-score (F1), and Matthews Correlation Coefficient (MCC).
RESULTS AND DISCUSSIONS
Performance comparison of PPIGAN with other approaches
In order to evaluate the performance of the proposed model, this article selects several representative methods as baselines and compares them with our method, all of which only utilize sequence features for PPI prediction. The description of these baseline methods is as follows:
In order to reduce the model bias and ensure fair comparison, these methods were validated on the same yeast dataset and the same human dataset. We extracted 20% of the samples from the whole dataset as the validation set for 5-fold cross-validation, and the negative samples in the validation set are all generated by the random sampling method. For the training dataset, the negative samples in PPIGAN are generated by the CGAN, whereas the negative samples in the other methods are generated by random sampling. The ratio of positive and negative samples in the training set or the validation set is 1:1. The comparison results of different models are shown in Tables 2 and 3.
Evaluation of PPI Prediction Performance on the Yeast Dataset Based on 5-Fold Cross-Validation
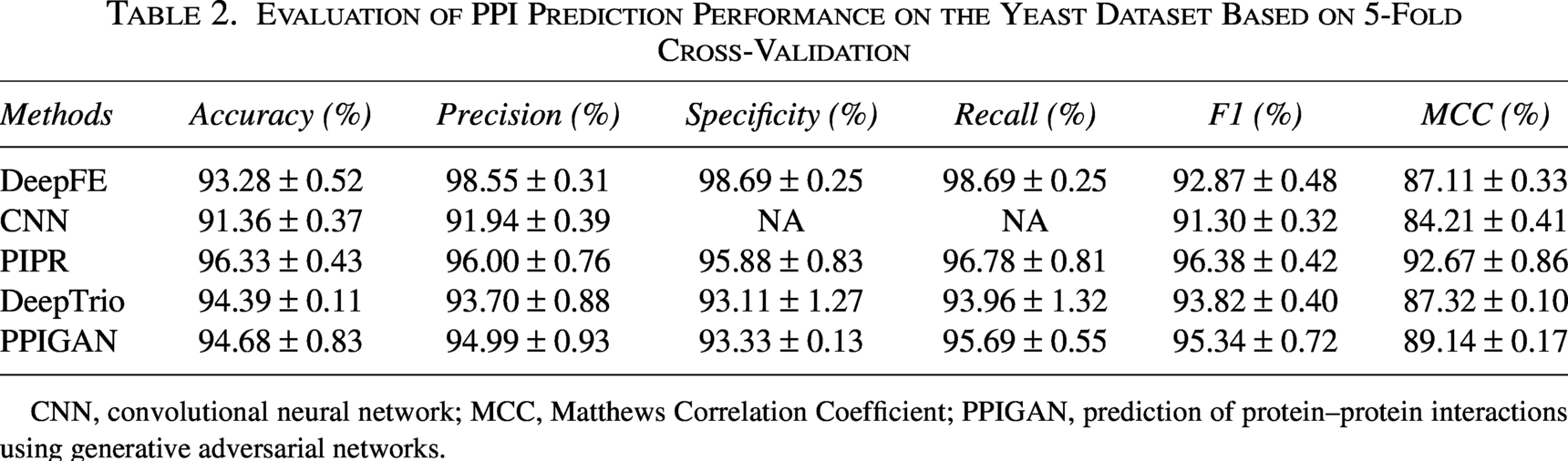
Evaluation of PPI Prediction Performance on the Yeast Dataset Based on 5-Fold Cross-Validation
CNN, convolutional neural network; MCC, Matthews Correlation Coefficient; PPIGAN, prediction of protein–protein interactions using generative adversarial networks.
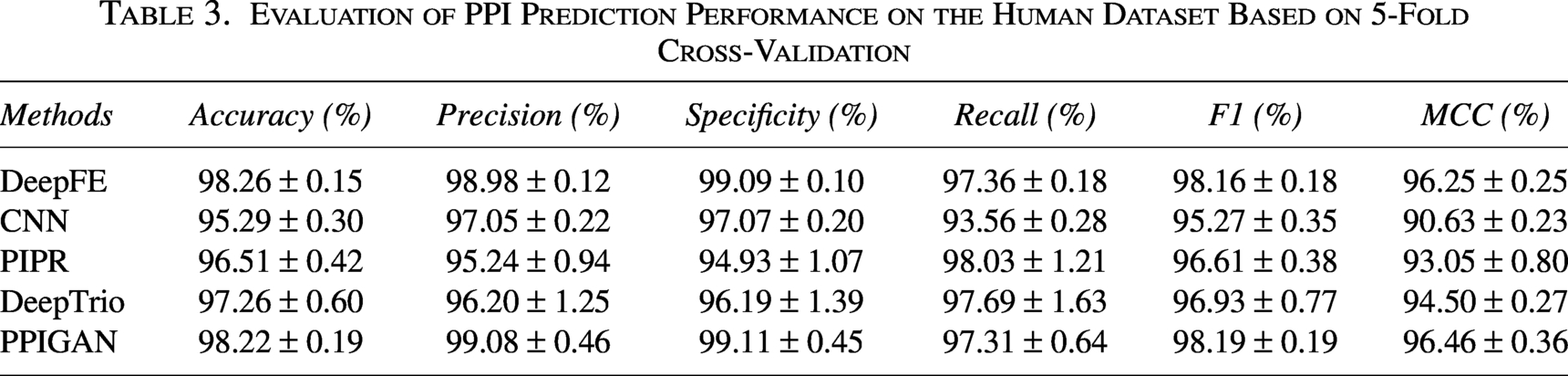
Evaluation of PPI Prediction Performance on the Human Dataset Based on 5-Fold Cross-Validation
As shown in Table 2, based on the 5-fold cross-validation results on the same yeast dataset, the proposed PPIGAN achieved competitive predictive performance. In terms of Accuracy, PPIGAN ranked second with 94.68%, which was 1.65% lower than PIPR but higher than DeepTrio and DeepFE by 0.29% and 1.40%, respectively. For Precision, PPIGAN (94.99%) was 3.56% lower than DeepFE but 1.29% higher than DeepTrio, and 1.01% lower than PIPR. PPIGAN also achieved an MCC value of 89.14%, ranking second after PIPR and demonstrating robust performance on this dataset.
As shown in Table 3, PPIGAN maintained robust performance on the human dataset. It achieved the best results in Precision, Specificity, F1, and MCC, and ranked second in Accuracy. In Accuracy, PPIGAN (98.22%) was 0.04% lower than DeepFE but outperformed DeepTrio and PIPR by 0.96% and 1.71%, respectively. For Precision and Specificity, PPIGAN exceeded DeepFE by 0.10% and 0.02%, respectively, and also consistently outperformed both DeepTrio and PIPR. In Recall, PPIGAN (97.31%) was slightly lower than PIPR, DeepTrio, and DeepFE by 0.72%, 0.38%, and 0.05%, respectively.
To evaluate the effectiveness of the GAN-based negative dataset proposed in this paper and compare predictive performance on different datasets, in addition to PPIGAN, two other methods of constructing negative datasets (2-let shuffling of positive sample sequences and random sampling) is used on the yeast data. The prediction models are DeepTrio and PIPR, and the prediction results are shown in Figure 4.
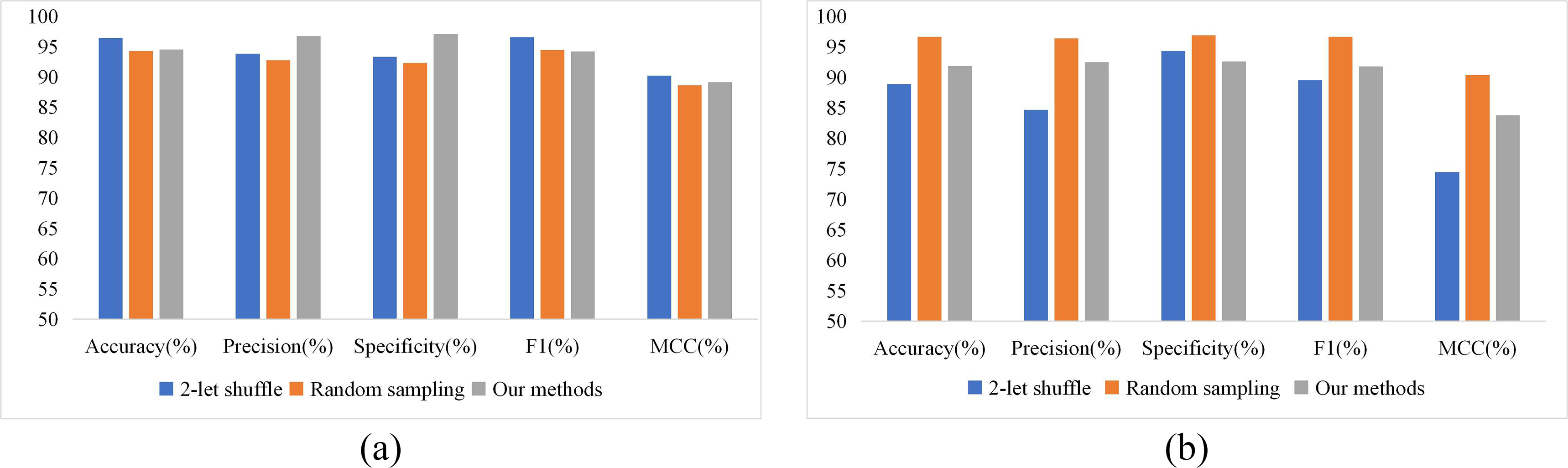
Performance of different negative sample construction methods.
As shown in Figure 4(a), the GAN-based negative sample construction method demonstrated strong performance in the DeepTrio model. It achieved particularly high scores in two key metrics: specificity (97.04%) and precision (96.71%), outperforming both the 2-let shuffling method (93.31%, 93.83%) and random sampling method (92.31%, 92.75%) by margins of 3.73–4.73% and 2.88–3.96%, respectively. While the accuracy (94.50%) was slightly lower than that of the 2-let shuffling method (96.42%), the MCC score (89.09%) confirmed the method’s robustness with imbalanced samples. Figure 4(b) reveals that when applied to the PIPR model, this method (91.87% accuracy) underperformed compared to random sampling (96.62%), but showed a 3% improvement over the 2-let shuffling approach (88.87%). Notably, it still maintained stable performance on the PIPR model with 92.47% precision and 92.59% specificity, demonstrating consistent reliability across different models.
In addition, in order to prove the validity of the generated protein sequences, the sequence alignment software Clustal 2.1 (Sievers and Higgins, 2021) was used to compare the generated sequences with the original sequences, and the visualization of two examples is shown in Figure 5. The software ESPript 3.0 was used to color the sequence comparison, where the red box indicates that the amino acids at the position are identical, and the blue transparent box indicates that the position is a conserved amino acid substitution. Sequence similarity was computed using BLAST 2.16 (default parameters) with local alignment and the BLOSUM scoring matrix. The reported 45% reflects the maximum similarity across all generated sequences when aligned to their native partner sequences (
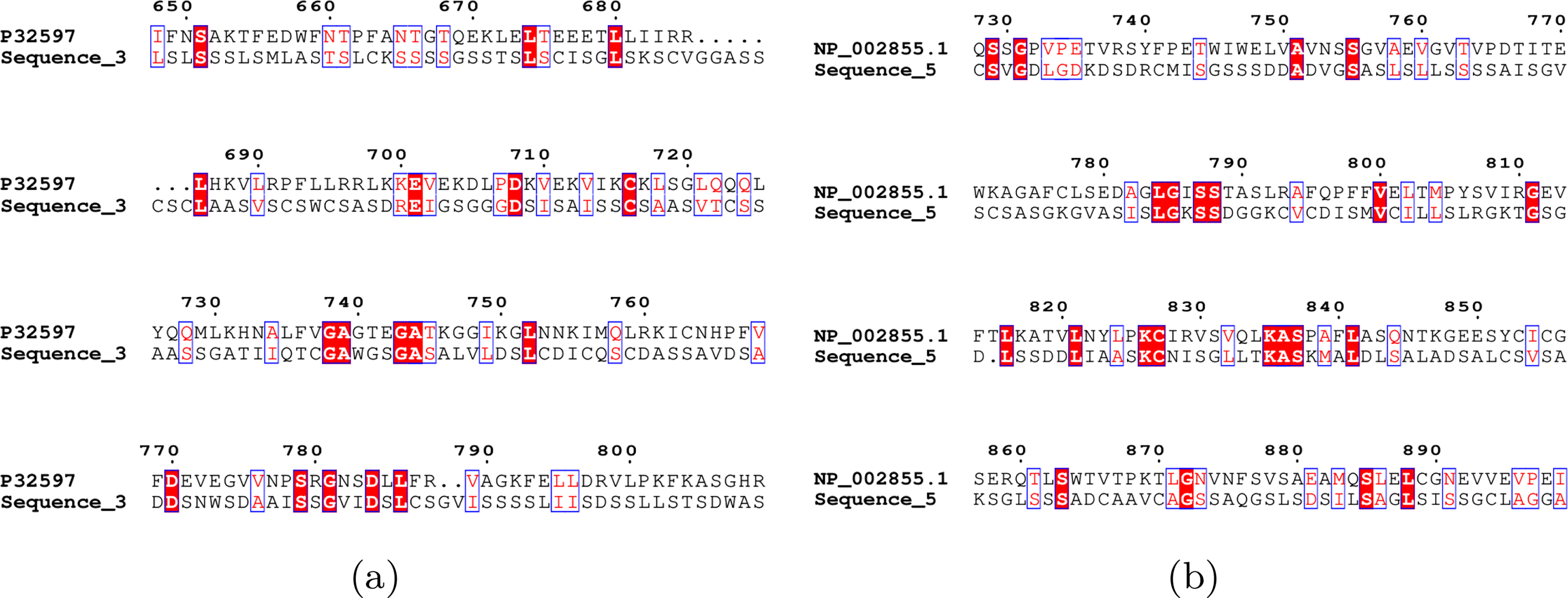
Alignment of the generated sequence with the original sequence.
To further evaluate PPIGAN’s performance in predicting unseen protein pairs, we employed an independent test set comprising human virus-protein interaction data from Wang et al. (2021), while training on the BioGRID human dataset (Hu et al., 2022) with a 1:1 positive-negative sample ratio. Again, we compared PPIGAN’s performance against both DeepTrio and PIPR, with test results displayed in Figure 6.
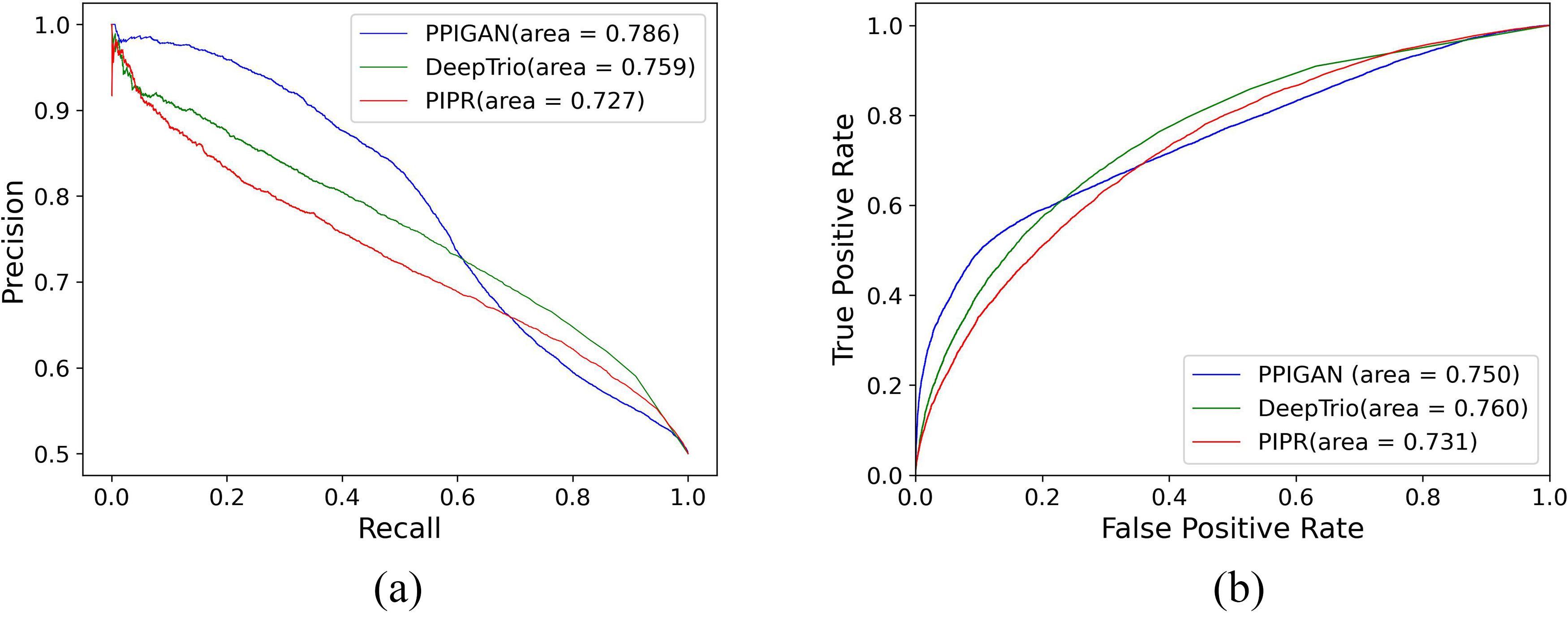
Performance comparison of PPIGAN with DeepTrio and PIPR on the independent test dataset.
As shown in Figure 6, for unforeseen human protein samples, PPIGAN demonstrated superior performance with an AUPRC (Area Under the Precision-Recall Curve) of 0.786, significantly outperforming both DeepTrio (0.759) and PIPR (0.727) (p-value < 0.0002). This indicates PPIGAN’s enhanced ability to balance precision and recall under imbalanced positive-negative sample conditions. The performance advantage likely stems from the generator’s degree centrality strategy, which produces more realistic negative samples (including interference pairs containing hub proteins), thereby enabling the discriminator to learn decision boundaries that better approximate the true data distribution. Furthermore, while PPIGAN’s AUROC (Area Under the Receiver Operating Characteristic Curve) (0.750) was marginally lower than DeepTrio’s (0.760) (p-value ≈ 0.011), it exhibited slower FPR growth in the low threshold range (FPR < 0.2), suggesting better control over false positive rates.
As mentioned earlier, our model achieved an accuracy of over 94% in cross-validation. To further verify its generalization ability, we conducted additional work in the following aspects:
Cross-species prediction
We trained the model on human PPI data (62,328 positive and negative samples) and tested it on yeast PPI data (11,188 positive and negative samples), and vice versa. In both scenarios, the accuracy was approximately 50%. Meanwhile, the other two methods (PIPR and DeepTrio) also achieved an accuracy of approximately 50% (see Supplementary Table S1, in which only the scenario of training on the yeast dataset and testing on the human dataset is presented). Additionally, we trained the model on human PPI data and tested it on mouse PPI data—sourced from the MINT database (Calderone et al., 2020), consisting of 10,861 positive samples and 10,861 randomly sampled negative samples-achieving an accuracy of around 54%.
We found that the prediction accuracy is related to the “similarity” between the training set and the validation set. For example, in cross-validation, nearly all proteins (99–100%) in the yeast test set had appeared in the training set, while an average of 71.7% of proteins in the human test set had appeared in the training set. In contrast, during cross-species prediction, proteins in the test set with high homology (sequence similarity > 40%) to those in the training set accounted for only approximately 44.17% (yeast vs. human) and 46% (mouse vs. human).
This indicates that the proposed model is highly valuable for predicting interactions between proteins that have already appeared in the training set. For instance, if interactions (p1, p2) and (p3, p4) are known, the model can effectively predict whether interactions (p1, p3) and (p2, p4) exist. However, it is less capable of predicting interactions between proteins that have not appeared in the training set or have low sequence similarity to proteins in the training set.
Test with no common proteins between training and test sets
For the previously used cross-validation dataset, we checked all proteins in the test set to see if they were present in the training set. If present, the corresponding protein pairs in the training set were removed. This ensured that the training set and test set had no common proteins, achieving “complete isolation.”
In this scenario, the number of positive samples in the training set decreased more significantly. The reason is that if a hub protein was identified in the test set, all protein pairs containing this hub protein in the training set were excluded, and we know that positive sample sets typically contain a higher number of hub proteins. Additionally, we further ensured that the sequence similarity between any two proteins in the dataset was <40% through clustering.
Under these conditions, the prediction accuracy was only approximately 50%. This indicates that the proposed model has limited predictive ability when there are no common proteins between the training set and the test set, and low homology among their proteins. This suggests, to some extent, that high homology between proteins is an important basis for PPI prediction, and hub proteins are also indispensable for PPI model training.
Testing on the Leakage-Free Gold-Standard Dataset
We performed further validation using the leakage-free gold-standard dataset established in (Bernett et al., 2024). In this dataset, the positive set was partitioned based on sequence overlap and homology to enforce low sequence similarity across the training, validation, and test splits, thereby preventing data leakage from identical or highly homologous proteins. Consistent with the “balanced random sampling” strategy proposed in Yu et al. (2010), the expected occurrence frequency of each protein in the negative set was matched to its node degree in the positive set, eliminating leakage caused by node-degree bias. On this gold-standard dataset, the accuracies of PIPR, DeepTrio, and PPIGAN ranged from 52.3% to 65.7%, with PPIGAN underperforming the other two models (see Supplementary Table S2). We hypothesize that this performance gap arises because PPIGAN trains on self-generated negative samples, whereas PIPR and DeepTrio were trained and evaluated entirely on the leakage-free positive and negative datasets.
Prediction on real negative sets
We selected two datasets provided by Negatome 2.0 (Blohm et al., 2014): the combined Manual-stringent and PDB-stringent dataset (6136 negative samples) and the standalone PDB-stringent dataset (4397 negative samples). Based on these two negative sets, we searched for corresponding positive samples in the MINT PPI database to ensure a large number of common proteins between the positive and negative datasets. Without sufficient common proteins, the positive and negative sets might become “far apart” due to no protein overlap, leading to artificially inflated accuracy. Using a sequence similarity threshold of 40% for clustering, we filtered to obtain low-similarity datasets.
The cross-validation accuracies were 81.67% (for the Manual-stringent and PDB-stringent dataset, with 1854 positive and negative samples) and 87.56% (for the PDB-stringent dataset, with 1968 positive and negative samples), respectively (see Supplementary Tables S3 and S4). On these two datasets, PPIGAN outperformed the other two models in terms of Accuracy, F1, and MCC. This shows that the proposed model can achieve reasonable prediction results on real negative sets.
The objective of this paper is to propose a new method for constructing negative datasets to improve the performance of protein-protein interaction (PPI) prediction models. We introduce a method, PPIGAN, based on CGANs, which iteratively generate negative samples that are sequentially similar to the positive samples, so as to form more sensitive and discriminative prediction models. The results demonstrate that the proposed negative sample construction method achieves competitive overall performance, and delivers significant improvement in the predictive performance of baseline models on real negative datasets.
In 5-fold cross-validation, PPIGAN achieved prediction accuracies of 94.68% and 98.22% on the yeast and human datasets, respectively, outperforming or matching other existing PPI prediction methods. Compared to the negative datasets generated by sequence shuffling and random sampling, the negative datasets generated by PPIGAN demonstrated the competitive prediction accuracy. On the independent test set, PPIGAN also achieved competitive performance compared with the other mainstream prediction methods (PIPR and DeepTrio), with superior performance on a subset of key metrics. Finally, we have identified the applicability and limitations of the PPIGAN model.
AUTHORS’ CONTRIBUTIONS
X.Z., S.X., and J.G. performed conceptualization, data curation, software development, formal analysis, and writing. X.W. joined part of data curation. L.L. performed part of conceptualization. L.H. provided partial computationalresources and constructive suggestions. J.Y. provided conceptualization, data curation, methodology, formal analysis, manuscript writing, and project supervision.
Footnotes
ACKNOWLEDGMENT
The authors thank the Artificial Intelligence Teaching Practice Platform of the College of Information Engineering and the High-Performance Computing Platform, both of Northwest A&F University, for providing computing resources.
AUTHOR DISCLOSURE STATEMENT
The authors declare they have no conflicting financial interests.
FUNDING INFORMATION
This work was supported by the grant of Key Research and Development Program of Shaanxi Province (Program No.
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.