
Abstract
Background
This paper describes and analyzes the cellular and molecular mechanisms underlying atherosclerosis development. In particular, the roles of monocytes/macrophages, smooth muscle cells, and vascular endothelium in the formation of stable/unstable atheromatous plaques, and the contributions of some processes to atheroma formation.
Methods and results
In this study we analyzed endothelium: function, dysfunction, and involvement into atherogenesis; cell proteins mediating mechanotransduction; proatherogenic role of monocytes; the role of macrophages in the development of unstable atheromatous plaques and smooth muscle cell origin in atherosclerosis. Smooth muscle cell phenotypic switching; their functioning; the ability to retain cholesterol and lipoproteins as well as secretion of pro- and anti-inflammatory molecules and extracellular matrix proteins, their response to extracellular stimuli secreted by other cells, and the effect of smooth muscle cells on the cells surrounding atheromatous plaques are fundamentally important for the insight into atherosclerosis molecular basis.
Conclusion
Atheromatous plaque transcriptome studies will be helpful in the identification of the key genes involved in atheroma transformation and development as well as discovery of the new targets for diagnosis and therapy.
Introduction
Cardiovascular disease (CVD) caused by atherosclerosis (AS) is the most common underlying cause of sickness and death in the world. Roth et al. 1 performed an analysis of the global rate of diseases and cardiovascular mortality from 1990 to 2015 and concluded that in 2015 every third death (17.92 million) was caused by CVD. According to the European Heart Network statistics, 2 CVD is the cause for 45% of all death cases in Europe, which is about 3.9 million deaths annually, and the total number of people suffering from CVD reached 85 million in 2015. Figures 1 and 2 respectively present the causes of death by rate among males and females, based on data collected for the latest available year in European countries.
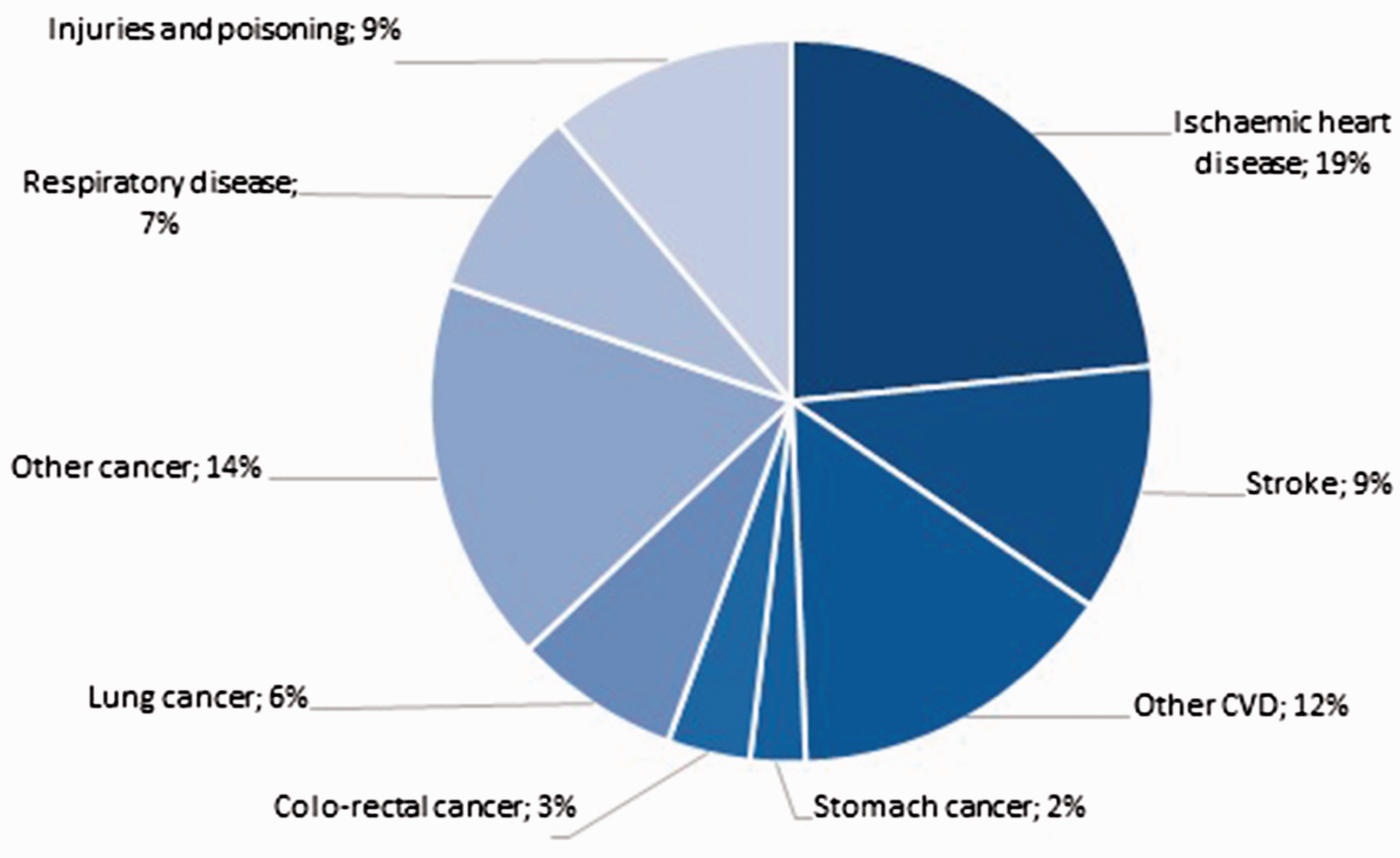
Causes of death by rate among males, Europe. 2

Causes of death by rate among females, Europe. 2
As seen from Figures 1 and 2, ischemic heart disease (IHD) and stroke are the most common forms of CVD. Each year in Europe IHD causes 862,000 deaths among men and 877,000 deaths among women; stroke is responsible for 405,000 and 583,000 death cases rateably. Further, according to the WHO estimates, 3 blood stroke heads the list of causes of the primary disability among adults (32 cases per 100,000 population). No more than 10–20% of the patients rehabilitate to normal activity after stroke with only 8% of them retaining their occupational fitness and 25% requiring attendance. 4 It should be noted that almost half (50–55%) of the ischemic stroke patients also suffer from atherothrombosis, which develops with the destruction of atheromatous plaque (AP) and forms a blood clot or an emboli in cerebral AS.
Nevertheless, it is an extremely widespread disease and neither are there efficient methods for AS treatment nor a reliable noninvasive AP diagnosis. The AS pathogenesis comprises a sequence of biological events in blood vessel intima, in particular, blood lipid/lipoprotein deposition, recruitment of monocytes/macrophages, foam cell formation, migration and proliferation of smooth muscle cells, secretion of extracellular matrix, and formation of the connective tissue in APs.5–7
The role of all the mentioned cell components in the development of AP stroma is quite uncertain, despite the consistent theory of AS pathogenesis. Currently, it is unclear which exactly cellular mechanisms or cells take part in the formation of slowly progressive stable APs (SAPs), neither they differences from the basic mechanisms of unstable APs (UAPs) development, displaying a rapid growth, trend to erosion, rupture of fibrous cap, release of atheromatous substance, and induction of local clotting. It should be noted that as much as 90% of all carotid thrombosis cases, which led to ischemic stroke, was caused by UAP rupture. The same statistic is applicable for acute coronary syndrome, acute myocardial infarction, and sudden cardiac death.8–14
In this article, we try to examine the cellular mechanisms of AS development, including the study of the role of monocytes, macrophages, and smooth muscle cells in the formation of stable/unstable APs. We believe that an understanding of these mechanisms is essential for the concept of AS pathogenesis, elaboration of precise AS diagnosis, and prediction of the disease course.
Function and dysfunction of endothelium and its influence on atherogenesis
Vascular endothelium (VE) plays an important role in the AP morphogenesis. It is the largest human endocrine organ (approximate weight is 2 kg in adult). 15 Beyond transcytosis (transport function), the endothelial cells also takes part in the implementation of a number of regulatory responses. One of the risk factors of the emergence of vascular diseases (e.g. AS) is a VE dysfunction. 16
Usually, there are two distinguished types of vascular endothelium namely atheroprotective and atherogenic, which are localized to different components of the cardiovascular system. 17 The atheroprotective VE has a profound glycocalyx layer; endothelial cells, regularly arranged and oriented along the blood flow, demonstrate a temperate proliferative activity. Typically, these cells have an ellipsoid shape (projection) and vast surface/length. 18 Glycocalyx is formed by adhesive oligosaccharide, polysaccharide, glycoprotein, and glycolipid molecules embedded into the plasmalemma. It is considered to provide receptor and marker functions and takes part in selective transport of substances through the endothelium. It is still unclear about the functional role of glycocalyx in atherogenesis; some scientists assume that its undamaged layer reduces the low-density lipoprotein (LDL) penetration into the subendothelial layer. 18
Atherogenic VE is characterized by chaotically arranged cubic cells with weakly outlined nuclei. 17 Endothelial cells located in atherogenic areas often have a weak barrier function and demonstrate high rates of proliferation and apoptosis. 19 Atherogenic VE retain ApoB-containing lipoproteins. 15 Besides, dysfunctional endothelium expresses proinflammatory cytokines and chemokines, extracellular matrix proteins, growth factors, and microRNAs, which are able to act as autocrine or paracrine factors enhancing further AS progression.19–23
There are several activators of endothelial dysfunction, such as mechanical activation, humoral factors, and activated immune cells. Due to its specific localization between blood and tissue, VE interacts with biologically active molecules, blood cells, and cells forming adjacent tissues. Moreover, cyclically changing hydraulic pressure of the blood and blood flow has a permanent impact on VE. Moreover, cyclically changing hydraulic pressure of the blood and blood flow has a permanent impact on VE. All of these factors can perform actions such as:
affect the processes in the nuclei and signal transduction pathways, activate or inhibit specific gene expression, stimulate or inhibit the synthesis of intracellular proteins, endothelial surface receptors, and biologically active molecules (proinflammatory cytokines, homeostatic factors, etc.) secreted by endothelial cells.
15
Because of the deformation of the endothelial membrane caused by blood flow, effects such as activating ion channels, changing glycocalyx and cytoskeletal proteins, and activating tyrosine kinase mechanoreceptors occur on the cell surface. It triggers intracellular signaling systems and synthesis of biologically active substances, which have a wide range of regulatory mechanisms (local and systemic). It should be noted that the type of endothelial response depends on the degree, direction, and constancy of shear stress, which is directly associated with the shear and flow velocity, blood viscosity, and vessel shape (curvature).
24
The mechanical forces acting on the vascular wall can be in the first approximation divided into two types:
radial forces emerging because of the blood pressure variation in systole/diastole; altering the vessel diameter, i.e. providing a pulse wave and shear forces (tangential forces), directed along the surface that emerge because of the blood flow along the vessel (in a more complex model, the superposition of these forces).
While the pulse wave systematically reduces from the center to periphery due to the expansion of the total cross-sectional area of the vessels, the radial force changes as well. The shear forces are primarily determined by the linear blood flow rate. The blood bed branches; correspondingly, the deformation caused by the mentioned forces and their superposition are nonuniformly spread along the blood bed.
25
Indeed, unbranched arteries with uniform blood flow have no predisposition to an atherosclerotic lesions development. And vice versa, the areas of major branching in the human carotid arteries (carotid sinus) are where early atherosclerotic changes appear.26,27
The mechanical influence can interfere with the gene expression profile. This is confirmed by both in vitro and in vivo observations. To assess the expression of the genes encoding glycocalyx components, cultivation of human endothelial cells under conditions imitating the mechanical stresses featured for atheroprotective and atherogenic regions of human carotid arteries was performed. This revealed the dependence of expression of some endothelial glycocalyx components (e.g. heparan sulfate proteoglycans and syndecan-1) on characteristics of shear stresses.28,29
Influence of blood flow on DNA methylation at the level of individual genes, as well as at global level, is confirmed by certain data. Indeed, a disturbance of normal blood flow leads to hypermethylation of the proximal promoter in Krüppel-like factor 4 (KLF4, belongs to the Krüppel-like transcription factor family, which takes part in embryonic development, cell proliferation, differentiation, and apoptosis) thereby preventing KLF4 transcription in the arterial regions with disturbed blood flow.30,31 Besides, other studies show a generalized effect that can have a blood flow through its impact on DNA methyltransferase 1 (DNMT1) activity,32,33 involved in supporting methylation (inhibition of this enzyme induces global DNA demethylation).
The NF-κB signaling pathway in endothelial cells is activated by atherogenic blood flow. 34 It causes an expression of proatherogenic receptors on the cell surface, including VCAM-1 and Toll-like 2 receptors. 35 Based on these data, it could be suggested that during the vascular system’s development the initial impact of mechanical forces on endothelial cells can “label” their DNA, which causes a special endothelial epigenetic landscape that depends on the flow.
Apparently, the type of endothelium phenotype (atheroprotective or atherogenic) could be determined by mechanically induced signaling pathways. Among these mechanisms, two antagonistic processes in endothelial cells play an important role: activation of the KLF2 and KLF4 transcription factors in atheroprotective endothelium, and activation of NF-κB signaling pathway in atherogenic endothelium.36–38 There are only a few genes that could be upregulated in endothelial cells and exposed to laminar shear stress, and some studies in vitro confirm that KLF2 is one of them.
39
The central signaling pathway for the activation of KLF2 and KLF4 expression is the MEK5/ERK5/MEF2 cascade. It can be modulated by:
AMP-activated protein kinase, siturin 1 protein, C-ζ protein kinase, SUMO-specific protease 2,
The importance of KLF2 for in vivo atheroprotection was demonstrated in a murine morel; in these experiments, Klf2+/–Apoe–/– mice displayed an increase in the atherogenic lesions in comparison with the control animal group, Apoe–/– mice.
16
Experiment with the Apoe–/– mice with KLF4 overexpression and knockdown confirmed the role of endothelial KLF4 in AS progression; its atheroprotective effect was undoubtedly clarified. 40 Numerous experiments also established an involvement of KLF2 in the regulation of wide range of genes controlling the endothelium functions, thrombosis, and hemostasis. Therefore, it can be concluded that it plays an important role in endothelial integrity and atheroprotection. 39
It was observed in endothelial cells that KLF2-dependent transcriptional and functional effects, exposed to atheroprotective flow, are quite similar to those that were exerted by statins. Gimbrone and García-Cardeña 39 investigated a possible connection between these biomechanical and pharmacological factors and found out that statins influenced the upregulation expression of KLF2. Mechanism of its influence is primarily connected with blocking of mevalonate production. Mevalonate produces isoprenoids such as farnesyl pyrophosphate and geranylgeranyl pyrophosphate (GGPP), which prenylate the distinct sets of proteins in the cells thereby providing their appropriate subcellular targeting and signaling. Depletion of GGPP explains the statin-mediated upregulation of KLF2. Note that from the point of view of its pro-inflammatory effect, statins make lesser impact on induction of KLF2 than the laminar shear stress.
The endothelial cells with an atherogenic phenotype promote the development of a proinflammatory environment, which is implemented via the NF-κB signaling pathway. NF-κB, the nuclear factor kappa B, is a major transcription factor responsible for cell adaptive response. NF-κB binds to promoter regions of over 100 genes (or 300 genes according to other data) responsible for inductive homeostasis. This factor plays an important role in cell proliferation, inflammatory response, and autoimmune reaction, since it regulates expression of the genes involved in these processes. The role of NF-κB signaling pathway at the initial stage of atherogenesis has been shown in several studies that demonstrate its activation in mouse aortic endothelial cells. 41
It is known that type 4 collagen and laminin are the major intercellular matrix proteins of blood vessels in the norm, whereas fibronectin accumulates in the atherogenic regions. In vitro experiments have allowed a research team to demonstrate that the exposure of endothelial monolayer to a turbulent flow upregulates fibronectin gene expression, thereby enhancing organization of atherogenic extracellular matrix and AP progression. 42 Some data suggest that disturbed hemodynamics can also influence mRNA splicing. For example, EIIA and EIIB exons of fibronectin, generally not expressed in the norm, are included by alternative splicing in the case of disturbed hemodynamics and regarded as a protective mechanism preventing aneurysmal dilatation of vascular wall. 43
Cell proteins and their influence on mechanotransduction
Mechanotransduction, which controls assembly and destruction of adhesive regions, is a mechanism that includes transduction of forces from outside via the cell–matrix and cell–cell contacts. A cascade signaling responsible for various changes in cell behavior is triggered in this process. Concentration of cytoskeletal elements in adhesion and intercellular contact sites causes the emerging of stress forces, which is the major inducer of mechanical signaling.
The mechanism of mechanotransduction in endothelial cells steel needs an elaborate understanding, despite the confirmation of this hypothesis by some recent experiments.44,45 Especially, the role of complex elements such as PECAM1, VE-cadherin, and VEGFR2 causing a change in endothelial phenotype exposed to mechanical stimuli was shown. Recent studies demonstrated that blood flow activates PECAM1-associated mechanotransduction via the GTP1 exchange factor, referred to as TIAM1, and activates Rac1 GTPase in endothelial cells. 46 The Rac1 activation triggers NF-κB signaling pathway and generation of reactive oxygen species.
The role of sphingosine-1-phosphate receptor 1 (a G protein-coupled receptor) in endothelial mechanotransduction is defined in other studies. 47 It demonstrates the importance of S1P1 expression in the arrangement of the cultivated endothelial cells along the flow and characteristic polarity of mouse descending aortic endothelium.
Piezo1 is another mechanoreceptor, mediator of shear stress, which should be studied in detail. Depending on the intensity of shear stress, it increases calcium flux, which initiates protease activities and partial remodeling of endothelial cell polarity. The endothelial cells isolated from Piezo1–/– mice failed to orient along the flow in response to blood flow impact. 48
Syndecan-4 is a transmembrane heparan sulfate proteoglycan. As has been revealed during the examination of shear stress along the blood flow in the mouse thoracic aorta, syndecan-4 participates in the endothelial cell alignment in culture. 49 Its deficiency combined with cholesterol diet led to the AS development in mice. The control group with normal syndecan-4 expression did not demonstrate such an effect.
Proatherogenic effect of monocytes
The immune cells that play a key role in the establishment of innate and acquired immunities are the monocytes. They are formed after the differentiation of progenitor bone marrow cells, accounting for 3–11% of the total leukocyte counts, and circulate for approximately 2–3 days after leaving the bone marrow to enter various tissues and transform into macrophages.
According to recent studies, disturbed cholesterol homeostasis can affect progenitor cells, manifesting in high counts of monocytes because of their overproduction. 50 It also became known that extramedullar myelopoiesis could occur under certain conditions and increase the monocyte counts in blood as well as in atherosclerotic lesions.51,52
Monocytes play an important role in AS pathogenesis. They are preactivated and show some characteristics of macrophages in peripheral blood of AS patients. Their adhesion to the endothelium is 1.5-fold higher as compared with the monocytes of healthy subjects; they express several receptors (types I and II Fcγ receptor and ICAM) and display elevated MHC II, characteristic of tissue macrophages. 53
The chemokine-induced monocyte trafficking from the bone marrow activates the endothelium in developing atherosclerotic lesions. Having entered the lipid- and lipoprotein-enriched intima regions, monocytes influenced by macrophage colony-stimulating factor (M-CSF), produced by activated endothelium, differentiate into macrophages; the macrophages actively phagocytize lipoproteins and transform into “foam” cells, loaded with lipids. Then macrophages consume lipoproteins, either native or modified by oxidation, aggregation, and/or other processes. As has been shown by in vitro experiments, lipoproteins can be processed via a combined pathway, namely, phagocytosis of aggregated lipoproteins, uptake of modified lipoproteins by scavenger receptors, and pinocytosis of native lipoproteins. 54
Macrophages form extracellular acid compartments for disaggregation/hydrolysis of aggregated LDLs. Having entered lysosomes, lipoproteins from these compartments are normally catabolyzed by hydrolases and lysosomal acids. A transient tight sealing of some regions in compartments is observed, allowing for holding “extracellular” proton gradient that enhances an increase in free cholesterol in these compartments. 55
Some data show that preservation and accumulation of lipoproteins in subendothelial space could contribute to AS pathogenesis. Mostly because it increases an expression of pro-inflammatory molecules and their receptors, which explains a long lead time of inflammatory response. 56 A murine AS model had demonstrated that this excludes potential inhibition of AP development by AS anti-inflammatory therapy. Indeed, the authors 57 succeeded in inhibiting AS in mice with the help of Ac2-26, a derivative of annexin A1.
Despite the evidence of proliferation of macrophages in the atherosclerotic lesions of arteries, it was commonly considered that each macrophage appears from one monocyte. Recent studies in murine AS models show that macrophage proliferation can be a quantitatively important stage in the macrophage accumulation in a full-scaled disease. 58 Experiments in vitro have shown that the activation of scavenger receptors and induction of phosphatidylinositol-3-kinase enhances macrophage proliferation. 59 The role of this mechanism in vivo is yet to be studied.
Advanced AS stage is characterized by chronic inflammation of AP macrophages, constant recruitment of monocytes, and efferocytosis. Chronic inflammation can correlate with some disturbance in inflammation inhibition. Normally, the inflammatory phase directly induces an anti-inflammatory response thereby enhancing the restoration of injured tissues and maintaining homeostasis. Some proteins (IL-10, TGF-β, and annexin A1) mediate the inhibition of inflammatory response as well as lipids (derivatives of arachidonic and omega-3 fatty acids, lipoxins, resolvins, protectins, and maresins).60,61
Mechanisms of macrophage activation in atherosclerotic lesions are quite certain for scientists; but modified lipoproteins and other damaging molecules are able to independently cause an activation of receptors involved in the inflammatory signaling, such as toll-like and nucleotide oligomerization domain (NOD)-receptors. Besides, the lipoprotein cholesterol accumulates in plasma membrane and alters the membrane characteristics, which boosts an inflammatory signal transduction from receptors.62,63 Oxidative stress inducing lipoprotein modification, oxysterol, and some other factors are also able to activate inflammatory processes. 64 Mitochondrial oxidative stress contributes to NF-κB activation with subsequent induction of the MCP-1 chemokine by monocytes and recruiting of monocytes. 65
The major source of local inflammatory mediators enhancing the AS progression are the macrophages. There are two currently distinguished main phenotypes of macrophages: “classic” or M1 phenotype (responding to intracellular pathogens) and “alternative” or M2 (responding to extracellular pathogens). It is known that part of monocytes can carry a preprogrammed ability to transform into M1 macrophages after entering an atherosclerotic lesion. 66
Activation of macrophage inflammatory signaling pathways is the critical proatherogenic process. As it was already mentioned, APs contain M1 (decisive in disease progression) and M2 (decisive in disease regression) macrophages, and the ratio M1/M2 can vary. The macrophage subpopulations with an M1-like phenotype additionally activate endothelium and increase monocyte recruitment. 67 Inflammasome induction plays an important role in the macrophage-mediated AS progression. It is a specialized protein complex formed in macrophages that triggers inflammatory processes when contacting with foreign proteins. Based on the results of various in vitro and in vivo experiments, it could be suggested that cholesterol microcrystals in macrophages can activate inflammasomes. 68 Other mechanisms potentially involved in triggering of inflammasome activation pathway in macrophages are also actively studied. Especially, the CD36 activation with the help of modified lipoproteins, which promotes transformation of such a cytoplasmic molecules as β-amyloid into inflammasome activating stimulus. 69 In addition, the mitochondrial DNA molecules oxidized by reactive oxygen species are also able to activate inflammasomes. 70
The proatherogenic effect of macrophages is realized through their interference with balance between inflammatory and anti-inflammatory responses. 54 In this process, lipid accumulation by foam cells does not always lead to inflammation. Indeed, desmosterol accumulation emerged by lipid metabolism induces such homeostatic processes such as activation of liver X receptors, reprogramming of fatty acid metabolism, and suppression of proinflammatory genes, observed in these cells. The macrophage activation in atherosclerotic lesions is most likely integrated with the induction of proinflammatory signals by the vascular wall. 71
The balance between synthesis of proinflammatory leukotriene B4 and anti-inflammatory lipoxin A4 in macrophages is another example of contribution made by two opposite processes. 72 Compensatory anti-inflammatory signaling pathways could be entailed by an inflammatory process. As an example, an acceleration of early AS progression in Ldlr–/– mice on a fatty diet caused by genetic block of the NF-κB signaling pathway, which can be associated with inhibition of IL-10 compensatory response. 73 Perhaps, simply blocking of just one of the possible pathways of inflammation development could not prevent AS progression because of its multifactorial nature. 74
Unstable APs and role of macrophages in its development
The subtype of atherosclerotic lesions that cause acute atherothrombotic vascular events are characterized by large necrotic areas and fibrous cap erosion. According to the current concept, macrophage apoptosis combined with defective efferocytosis (apoptotic cell clearance) leads to postapoptotic plaque necrosis. 75 RIP3 (receptor-interacting protein kinase 3), a protein inducing macrophage necrosis with subsequent inflammation induction, and primary necrosis mediated by this protein can also enhance AP rupture. Macrophages are able to aggregate into the so-called necrotic cores, releasing their content when the UAP cap is ruptured. 76
An increase of apoptosis can be caused by various factors, such as:
oxidized lipoproteins, oxidized phospholipids, lipoprotein overaccumulation in the endoplasmic reticulum, systemic risk factors associated with insulin resistance, direct impact of defective insulin signals on macrophages.
77
As it has been shown by in vivo experiments with a murine model, a C/EB homologous protein (CHOP) is an effector of endoplasmic reticulum stress and macrophage apoptosis inducer in AS.
78
Furthermore, a clear correlation between CHOP expression, apoptosis, and degree of AP instability in human coronary and carotid arteries is observed.79,80 CHOP induces different pathways of apoptosis activation. The most typical for macrophages is the activation of inositol-3-phosphate receptors, which involve in endoplasmic reticulum calcium release.
81
An increase in cytosolic calcium induces expression of Fas receptors via the CaM-kinase IIγ (calcium/calmodulin-dependent protein kinase IIγ) signaling pathway and concurrently triggers the mitochondrial apoptosis pathway.
82
In addition, CHOP inhibits expression of Bcl-2 (an intracellular protein factor) family proteins. Bcl-2 inhibits apoptosis in many cell systems (including lymphohematopoietic cells) and regulates cell death by controlling the mRNA membrane permeability. A downregulation of Bcl-2 expression enhances death of all Bcl-2-deficient macrophages and development of AP necrosis.
Macrophage efferocytosis is defective in the advanced AS stage. Efferocytosis is implemented via the interaction between apoptotic signals on the surface of apoptotic cells and macrophage receptors. 83 MerTK (Mer tyrosine kinase) is regarded as a putative receptor for apoptotic molecules. 84 It is exposed on the macrophage surface, which is functionally important for efferocytosis and subject to ADAM17-mediated hydrolysis. MerTK hydrolysis gives Mer-soluble peptide, which acts as a competitive inhibitor for efferocytosis implemented by blocking ligands on the apoptotic cell surface. Indeed, MerTK hydrolysis has been observed in developing APs, especially in necrotic UAPs. 85
CD47 is a key molecule blocking phagocytosis, and its upregulation is associated with atherogenesis. 86 The experiments with a murine model shows that intravenous administration of anti-CD47 antibodies halves the lesion area.
Along with the disturbance of efferocytosis, macrophages with the help of metalloproteinase secretion are likely to enhance fibrous cap thinning and stabilize APs. However, this is almost impossible to prove in vivo because murine AS models cannot simulate such features of human UAPs as AP rupture. 87
Smooth muscle cell origin in atherosclerosis
The main function of SMCs (highly specialized and differentiated cells of the body) is contraction. It allows regulation of vascular tone and diameter, thereby controlling arterial tension and blood flow distribution. SMCs in blood vessels proliferate at an extremely low rate and display a very low synthetic activity. These cells express a unique set of contractile proteins, ion channels, and signaling molecules necessary for implementation and regulation of their contractile function.
An activation of SMC and the change in their phenotype is a result of the influence of factors such as lipoprotein accumulation, activation of endothelium, and inflammatory response in the development of atherosclerotic lesion. The resting SMCs are typical in their phenotype downregulate expression of SMC differentiation gene markers, encoding smooth muscle α-actinin (Acta2), and myosin heavy chains (Myh11). SMCs commence active proliferation, migration, and production of extracellular cell matrix, proteoglycans, which are proteins remodeling blood vessels and stabilizing APs. 6
There are various factors that initiate the switching of SMC phenotype. The results of in vitro experiments have shown that platelet-derived growth factor (PDGF) stimulates SMC migration and proliferation. Also, certain data collected in vivo allow to suggest that PDGF enhances AP growth at the expense of SMCs. Presumably, PDGF enhances SMC proliferation and AP fibrous cap formation.88,89 Transforming growth factor beta (TGF-β) is a strong stimulant for the production of SMC intercellular matrix, in particular, collagen IV, leading to SMC matrigenic (synthetic) phenotype. On the other hand, oxidized phospholipids are likely to induce the SMCs of an intermediate phenotype by stimulating SMU migration and proliferation as well as upregulation of type VIII collagen expression.90,91
Inflammatory cytokines such as tumor necrosis factor alpha (TNFα) and interleukin-1 (IL-1) stimulate ICAM-1 (intercellular adhesion molecule 1) expression in SMCs as well as IL-6 and metalloproteinases, which in turn form SMC inflammatory status. 92 The SMCs-initiated ICAM-1 expression enhances monocyte/macrophage and T-cell adhesion through the binding of lymphocyte function-associated antigen (LFA-1), expressed on the surface of the cells with ICAM-1. 6 In addition, in vitro experiments have shown that an elevated inorganic phosphate concentration gives rise to osteochondrogenic precursors. As a consequence, it increases the calcification, and it is possible to observe the SMCs of this type in calcified blood vessels of matrix Gla protein-deficient mice.93,94
The UAPs are the most suitable for defragmentation and are responsible for thromboembolic events, which contain less SMCs in comparison with foam cells. However, it is still unclear as to which part of AP cells originates from SMCs and which part from monocytes. Some articles demonstrate first results. Notably, SMCs markers such as smooth muscle Acta2, myosin heavy chains, and SM22α/transgelin in the developed APs of Apoe–/– mice, prone to AS, display a low expression level. 95 Moreover, SMC cultivation in a cholesterol-loaded medium downregulates SMC marker gene expression and induces macrophage markers, including CD68 and Mac2. 6 Presumably, this phenomenon also takes place in APs: the experiments with genetically marked SMCs of Apoe–/– mice have shown that the cells with a smooth muscle origin in the developed APs express Mac2 and CD68; but only 11% rate of labeling efficiency could not allow to estimate the overall SMC contribution to AP formation. 96 The function of these cells in AS pathogenesis is still unclear and require a clarification: are they simply present in AS lesions or play an important role in the AP formation and development.
The macrophages or at least hematopoietic cell derivatives are also able to express SMC markers, including smooth muscle α-actin and SMА2α. In particular, the cultivated macrophages treated with TGF-β or thrombin express SMC markers and the studies tracking the cell lineages suggest that hematopoietic cell derivatives in Apoe–/– mice express SMC markers at the early (but not late) stages of their differentiation.97,98 Later experiments confirm the local origin of almost all cells, which are positive for smooth muscle markers in the atherosclerotic lesions of Apoe–/– mice.99–101 Genetic tracking according to Y chromosome markers in the sex-mismatched bone marrow transplant subjects demonstrate that over 10% of the α-actin positive cells in developed APs in diseased coronary arteries are of a hematopoietic origin rather than smooth muscle origin.102–103
Conclusions
The understanding of AS progression is impossible without the research of AP cells and their response to external stimuli. For example, the KLF4 encoding gene acts as an atheroprotector, but at the same time it is able to reduce the AP stability because its expression in SMCs can decrease the number of SMCs. Deletion of KLF4 in SMCs allows to demonstrate that this factor is responsible for phenotypic switching of cultivated SMCs from a contractile to a synthetic phenotype in response to PDGF-BB treatment or oxidized phospholipids. On the contrary, IL-1β expression in macrophage-like cells indicates their proatherogenicity, whereas this expression in SMCs suggests AP stability.
These examples demonstrate how different cells respond to the ambient stimuli and emphasize the importance of comprehensive phenotype determination of the cells expressing particular genes. With regard to SMCs, there are few essential points for better understanding of AS molecular basis:
phenotypic switching; functioning; ability to retain cholesterol and lipoproteins; secretion of pro- and anti-inflammatory molecules and extracellular matrix proteins; response to extracellular stimuli secreted by other cells; influence on cells surrounding APs.
Evidently, AP transcriptome studies will be helpful in the identification of the key genes involved in atheroma transformation and development as well as discovery of the new targets for diagnosis and therapy.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Russian Science Foundation (Grant No. 18-75-10030).