
Abstract
Objective
Peripheral artery disease patients have been shown to be more susceptible to thrombotic events compared to non-peripheral artery disease patients. Therefore, the aim of this study was to investigate the coagulation profile in peripheral artery disease patients with chronic limb threatening ischemia, moderate peripheral artery disease patients with claudication, and non-peripheral artery disease controls.
Methods
Chronic limb threatening ischemia patients were matched to peripheral artery disease patients with claudication and non-peripheral artery disease controls in a 1:1:1 ratio. Each patient had their cytokines, markers of thrombin generation, coagulation factors, natural anti-coagulants, fibrinolysis, and endothelial injury markers assessed.
Results
Markers of thrombin activation, thrombin Fragments F1 + 2 (Frag 1 + 2), and thrombin–anti-thrombin complex were found to be significantly elevated in all peripheral artery disease and chronic limb threatening ischemia patients relative to non-peripheral artery disease controls. Similarly, relative to non-peripheral artery disease controls, inflammatory markers including C-reactive protein, soluble platelet factor 4, and neutrophil gelatinase-associated lipocalin were also found to be significantly upregulated in chronic limb threatening ischemia patients, but not in peripheral artery disease patients with claudication. Furthermore, our data demonstrated significant increases in markers of endothelial injury in chronic limb threatening ischemia patients relative to non-peripheral artery disease controls. Finally, decreases in natural anti-coagulants (protein C and protein S) and coagulation factors FIX, FXI, and FXII were also observed in chronic limb threatening ischemia patients when compared with non-peripheral artery disease controls.
Conclusions
Our data suggest that in relation to non-peripheral artery disease controls, chronic limb threatening ischemia patients are more hypercoagulable. However, peripheral artery disease patients with claudication appear to have similar levels of circulating procoagulant markers as non-peripheral artery disease patients. This may explain the increased risk of thrombotic events observed in chronic limb threatening ischemia patients.
Keywords
Introduction
Lower extremity peripheral artery disease (PAD) is a common presentation of atherosclerosis, with over 200 million people reportedly living with the disease.1–3 It is estimated that the global prevalence of PAD is 29% in individuals with cardiovascular risk factors. 4 Furthermore, PAD has a broad spectrum of clinical presentations. 5 For instance, the majority of PAD patients are asymptomatic. 4 In contrast, symptomatic patients often present with claudication, defined as muscular pain with ambulation, with a minority of PAD patients presenting with a threatened limb. 6
Symptoms induced by PAD are usually a result of arterial stenosis, occlusions, and thrombotic events secondary to plaque rupture.7,8 From our current understanding of plaque pathophysiology, ruptured plaque results in the exposure of endothelial thrombogenic proteins that promotes the activation of circulating coagulation factors leading to the activation of the coagulation system and thrombus formation within the vessel. Hence, this is postulated to result in disease progression.9,10 Patients who suffer from PAD are classified as per the Rutherford Classification of chronic limb ischemia based on their clinical status into three major groups: (1) asymptomatic (stage 0), (2) patients with claudication (stages 1–3), and (3) chronic limb threatening ischemia (CLTI) (stages 4–6). 11 It is estimated that over a period of five years, 5%–25% of PAD patients experience disease progression to CLTI, the most severe form of PAD.12–15
To date, the relationship between advancing PAD severity and thrombotic state has not been fully understood, but studies suggest that CLTI patients have increased circulating levels of markers indicative of a prothrombotic state in relation to patients without PAD.7,8,16 Therefore, in this pilot study, we sought to better understand the factors involved in coagulation system activation by comparing the levels of various regulatory proteins and coagulation factors in CLTI patients and PAD patients (with claudication) versus non-PAD controls.
Patients and methods
Ethics approval
This study was approved by the research ethics board at St. Michael's Hospital—University of Toronto in Ontario, Canada. The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki, and informed consent was obtained (in written) from all participants.
Patient selection
The PAD status was defined clinically as per the Rutherford Classification Criteria of chronic limb ischemia. 11 In all subjects, a thorough physical exam and complete medical history was obtained by an independent PAD expert. Arterial ultrasound (US) findings including ankle brachial index (ABI) were recorded for each patient. Patients with CLTI (Rutherford stage ≥4, ABI <0.9 or brachial index (TBI) <0.67 with evidence of rest pain or tissue loss) referred to vascular surgery ambulatory clinics or the emergency department at St. Michael’s Hospital between March 2018 and May 2018 were asked to participate. We excluded the following: (1) patients on anticoagulants, chemotherapy, or biological anti-inflammatory agents, (2) patients diagnosed with acute stroke or transient ischemic attack (TIA) within six months, (3) patients diagnosed with sepsis or deep vein thrombosis, (4) patients with history/active cancer, and (5) patients with acute or six-month history of acute coronary syndrome (ACS), heart failure, or uncontrolled arrhythmia as defined by American College of Cardiology.17–22 A total of 247 consecutive patients were considered for this study; however, only 20 CLTI patients met the inclusion criteria and consented to participate. Next, the CLTI patients were matched with PAD patients with claudication (defined as Rutherford stage 1–3, ABI <0.9 or TBI <0.67 with the absence of rest pain or tissue loss) and non-PAD controls (ABI >0.9, TBI >0.67 and no clinical evidence of PAD along with palpable distal pulses) in a ratio of 1:1:1. Patients were matched by age group, sex and history of coronary artery disease, stroke, hypercholesteremia, hypertension, active smoking, and diabetes. To do so, during the months of June–July 2018, 20 PAD patients with claudication and 20 non-PAD controls were recruited to match the CLTI cohort.
Baseline measurements and sample processing
Medical history including details of any previous ACS, hyperlipidemia, arterial arrhythmia, arterial hypertension, renal disease, congestive heart failure, history of stroke or TIA, history of cancer, diabetes, and smoking status. Each patient received lower limb arterial imaging (arterial US, computed tomography angiogram (CTA) or angiogram) as part of the PAD assessment. For patients with non-compressible ABI (ABI >1.3), the TBI was used to identify PAD status. A TBI <0.67 was considered abnormal.
Blood samples were drawn into vacutainer tubes containing EDTA. Plasma was then prepared from the anti-coagulated blood via centrifugation at 1000 g for 10 min (4°C), which was then aliquoted and stored at −80°C. Plasma samples that had previously been thawed were not utilized for this study.
Biomarkers of thrombin activation and inhibition analysis via ELISA assays
Biomarkers indicative of thrombin activation were measured in control and experimental patients. Pro-thrombin fragments 1 + 2 (Frag 1 + 2), (Cedarlane, Massachusetts, USA) and thrombin–anti-thrombin complex (TAT), (Abcam, Cambridge, UK) were measured via ELISA. Optical density was determined using a microplate reader. Duplicate assays were performed for each sample, and the plasma optimal dilution factor was identified for each tested biomarker within both assays. All sample analysis for each protein was completed on the same day to eliminate inter-assay variability. Both sample intra-assay and inter-assay CV were <10% for all biomarkers.
Protein multiplex assays
Cytokines, coagulation factors, and natural anti-coagulants were measured in all patients. Plasma samples were analyzed in duplicate using Coagulation Human ProcartaPlex™ Panels 1 and 3 (Thermo-Fisher, Massachusetts, USA) to determine the antigenic concentrations of Factor XI, Factor XII, Factor XIII, antithrombin, C-reactive protein (CRP), Factor IX, protein S, and protein C. Inflammatory cytokines, platelet activation biomarkers, regulators of coagulation cascade, and endothelial cell injury markers were also investigated in each patient using multiplex kits from Millipore Sigma (Burlington, Massachusetts, USA). The human cardiovascular 4 MAG cardiovascular kit was used to analyze thromobomodulin, tissue factor, Pentraxin 3, and E-selectin, while the human cardiovascular 2 MAG cardiovascular kit was used to measure the levels of D-Dimer, sICAM-1, growth differentiation factor-15 (GDF 15), P-selectin, neutrophil gelatinase-associated lipocalin (NGAL), and sVCAM-1. Finally, the human cardiovascular 3 MAG kit was used to measure the levels of alpha-1-acid glycoprotein (AGP), L-selectin, fibrinogen, and PF4. For all multiplex studies, analysis was completed as described by the manufacturer. Sample intra-assay and inter-assay CV were both <10%. The MagPix analyzer (Luminex Corp; Austin, Texas) was calibrated prior to analysis using Fluidics Verification and Calibration bead kits (Luminex Corp). A minimum of 50 beads for each targeted biomarker were acquired using Luminex xPonent software and analyzed using Milliplex Analyst software (v.5.1; EMD-Millipore).
Statistical methods
SPSS software, version 23 (SPSS Inc., Chicago, IL, USA) was used for data entry and analysis. All analyses were carried out at a 5% two-sided significance level. Demographics and baseline measurements were recorded for each patient. Continuous variables were tested for normality using Shapiro–Wilk test and normality plots. Normally distributed continuous variables were summarized and reported in terms of means as well as standard deviation. For non-normally distributed data, the median and interquartile ranges (IQR) were calculated. Finally, categorical variables were reported as percentages. Evaluation of baseline characteristics was done using independent t tests or Mann–Whitney U test for continuous variables. Fisher’s exact test or chi-square test was used for categorical variables. Sample matching was done using SPSS case–control matching method. Each CLTI case was matched with non-PAD case and PAD case with claudication in a ratio of 1:1:1. Since the studied protein concentrations failed normality assumption, comparisons of independent samples were done using Mann–Whitney U test, while Kruskal–Wallis test was used to compare more than two independent groups. Post hoc test was then used for pairwise comparisons to further analyze these differences.
Results
Patients
We recruited a total of 60 patients (20 CLTI patients matched to 20 PAD patients with claudication and 20 non-PAD patients). Between the three groups, mean age was between 64 and 67 years old. The majority of participants (65%) were males in all three groups, and prevalent comorbidities include diabetes, hypertension, smoking, and hypercholesterolemia, among others. Naturally, the ABI and TBI were significantly different between all three groups. None of the enrolled patients had a prior history of revascularization intervention (either surgical or endovascular) for PAD. Clinical characteristics of the participants are shown in Table 1. Figure 1 demonstrates all the different protein markers that affect the coagulation cascade which were investigated as part of this study.
Baseline characteristics for the patient cohort by subgroup (N = 60).

ACEi/ARB: angiotensin converting enzyme inhibitor/angiotensin-receptor blockers; non-PAD: none peripheral arterial disease; CLTI: chronically limb threatening ischemia; Cr: creatinine; WBC: white blood cell; HbA1C: hemoglobin A1chemoglobin A1c; HDL: high density lipoprotein; HbA1C: hemoglobin A1c; LDL: low density Lipoprotein; ABI: ankle brachial index; TIA: transient ischemic attack.
Rutherford Classification of Chronic limb ischemia: claudication (stages 1–3); and chronical threatened limb ischemia (stages 4–6).
aContinuous variable summarized as means and standard deviations. ANOVA was used for comparing means. p value represents comparison across the three study groups.
bCategorical variables summarized by frequencies and percentages. Chi-Square test was used to compare percentage differences between groups. p value represents comparison across the three study groups.
All mean, standard deviation, and median values as well as their measures of variability were formatted to one decimal place. All percentages were rounded to one decimal place. All hypothesis testing was carried out at the 5% (two-sided) significance level. All p values were presented with at least three decimal places, and p values less than 0.001 were reported as <0.001 in tables.

Crosstalk between the coagulation cascade and investigated procoagulant markers.
Evidence of coagulation cascade and platelet activation in CLTI patients
To examine whether CLTI patients are more prone to thrombosis, we measured the plasma levels of two markers indicative of thrombin activation: TAT and pro-thrombin fragments 1 + 2 (Frag 1 + 2). Since platelets play a major role in clot formation, we also measured the plasma levels of soluble platelet markers that are indicative of platelet activation including: P-Selectin, platelet factor 4 (sPF4), and VCAM1 (sVCAM1). Finally, we measured D-dimer, which is indicative of fibrinolysis. As illustrated in Table 2, there was a statistical difference in circulating levels of all studied markers in the CLTI group relative to non-PAD controls. When the less severe PAD patients with claudication were compared to non-PAD controls, none of the investigated markers were elevated. When all PAD patients (Rutherford ≥1 and ABI <0.9) were compared to non-PAD controls, a significant increase was only noted for P-Selectin, sVCAM-1, VWF, and TAT. These data indicate that relative to non-PAD patients, the coagulation system is activated to a higher intensity in patients with CLTI but not in PAD patients with claudication. Also, despite being on anti-platelet medication, our data show that patients with CLTI had significantly higher levels of platelet activation markers relative to control patients.
Comparison of coagulation and platelet activation markers between non-PAD controls and patients with increased severity of PAD.

PAD: peripheral artery disease; CLTI: chronically limb threatening ischemia; TAT: thrombin-anti-thrombin complex; PF4: platelet factor 4.
Non-PAD group (no clinical symptoms of PAD, palpable distal pulses, ABI >0.9 and TBI >0.67) n = 20; PAD with claudication (Rutherford 1–3, absence of rest pain or tissue loss, ABI <0.9 or TBI <0.67) n = 20, and CLTI (Rutherford ≥4 with presence of rest pain or tissue loss, ABI <0.9 or TBI <0.67) n = 20.
Fold-change was a ratio calculated based median protein concentration levels.
aRutherford stage ≥1.
bKruskal–Wallis test post-hoc results for CLTI vs non-PAD and Claudication vs non-PAD.
cMann–Whitney test represents the difference in protein concentrations between PAD and non-PAD patients.
dDenotes soluble form of the investigated protein.
Increase in inflammatory response and endothelium injury in CLTI patients
Since endothelial injury and inflammation are known to promote coagulation cascade activation, 23 we were keen on learning the relationship between these pathways in CLTI, claudicants, and non-PAD controls. To do so, we measured the levels of circulating cytokines which are reported to be increased in patients with cardiovascular disease. In this study, the following inflammatory markers were investigated: CRP, AGP, 24 GDF-15, 25 soluble L-selectin, 26 NGAL, 27 and pentraxin-3 28 (Table 3). We also measured the following soluble forms of endothelial proteins, which are released into circulation post endothelial injury: tissue factor (sTF) and thrombomodulin (sTM), sE-selectin, and sICAM-1. Once again, relative to non-PAD controls, there was a significant increase in the levels of AGP, GDF-15, L-selectin, NGAL, sICAM-1, TM, and TF in CLTI patients and not in patients with claudication (Table 3). These results demonstrate an elevation in the inflammatory response as well as increased evidence of endothelial injury as the severity of PAD increases. Finally, when the non-PAD controls were compared to all PAD patients, we noticed a significant difference in the levels of AGP, L-selectin, GDF-15, NGAL, sICAM-1, and sTF. This indicates that when we group all PAD patients together, the elevation in inflammatory and endothelial injury markers were mainly driven by the CLTI cohort.
Comparison of inflammatory and endothelial injury markers between non-PAD controls and patients with increased severity of PAD.
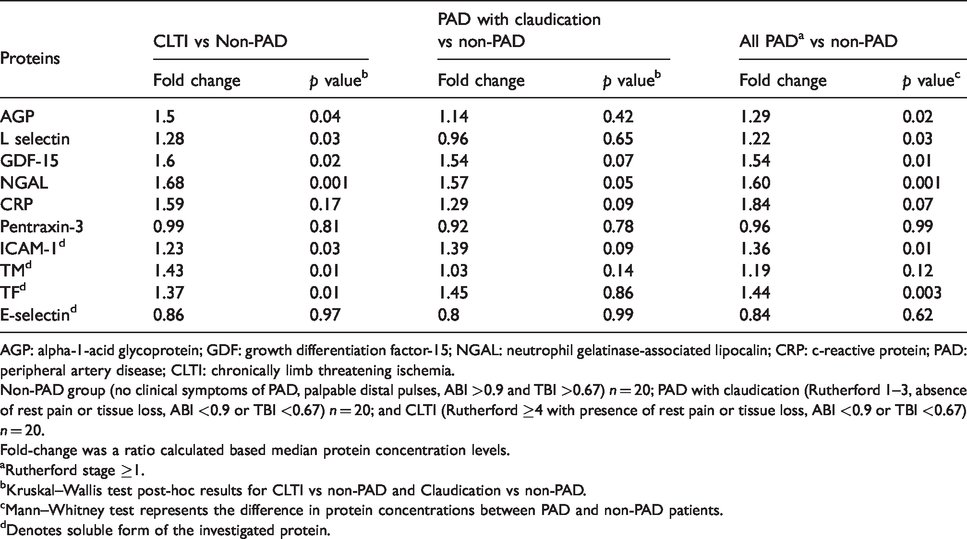
AGP: alpha-1-acid glycoprotein; GDF: growth differentiation factor-15; NGAL: neutrophil gelatinase-associated lipocalin; CRP: c-reactive protein; PAD: peripheral artery disease; CLTI: chronically limb threatening ischemia.
Non-PAD group (no clinical symptoms of PAD, palpable distal pulses, ABI >0.9 and TBI >0.67) n = 20; PAD with claudication (Rutherford 1–3, absence of rest pain or tissue loss, ABI <0.9 or TBI <0.67) n = 20; and CLTI (Rutherford ≥4 with presence of rest pain or tissue loss, ABI <0.9 or TBI <0.67) n = 20.
Fold-change was a ratio calculated based median protein concentration levels.
aRutherford stage ≥1.
bKruskal–Wallis test post-hoc results for CLTI vs non-PAD and Claudication vs non-PAD.
cMann–Whitney test represents the difference in protein concentrations between PAD and non-PAD patients.
dDenotes soluble form of the investigated protein.
Comparison of natural anticoagulants and clotting factors between non-PAD controls and patients with increased severity of PAD.
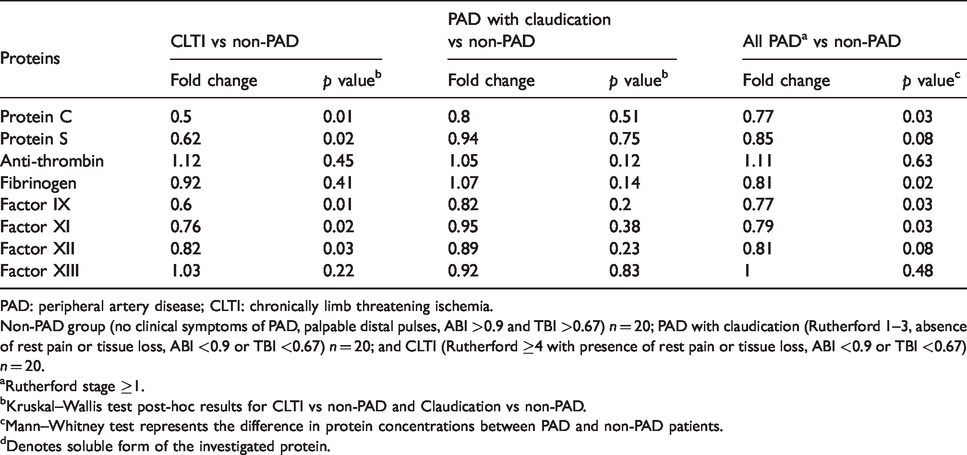
PAD: peripheral artery disease; CLTI: chronically limb threatening ischemia.
Non-PAD group (no clinical symptoms of PAD, palpable distal pulses, ABI >0.9 and TBI >0.67) n = 20; PAD with claudication (Rutherford 1–3, absence of rest pain or tissue loss, ABI <0.9 or TBI <0.67) n = 20; and CLTI (Rutherford ≥4 with presence of rest pain or tissue loss, ABI <0.9 or TBI <0.67) n = 20.
aRutherford stage ≥1.
bKruskal–Wallis test post-hoc results for CLTI vs non-PAD and Claudication vs non-PAD.
cMann–Whitney test represents the difference in protein concentrations between PAD and non-PAD patients.
dDenotes soluble form of the investigated protein.
Decrease in natural anti-coagulants and clotting factors with PAD
Natural anti-coagulants play an important role in maintaining a hemostatic balance of clot formation by inhibiting activated coagulation factors. 29 Any disturbance in the level of such regulators will have a role in the pro-thrombotic state. To investigate the role of natural anticoagulants and clotting factors in PAD patients, we measured the levels of protein C, protein S, anti-thrombin as well as the circulating levels of FIX, FXI, FXII, FXIII, and fibrinogen in CLTI, claudicants, and control patients. We observed a significant decrease in the levels of protein C, protein S, Factor IX, Factor XI, and Factor XII in CLTI patients relative to non-PAD controls. This was not the case when claudication patients were compared with non-PAD controls. Interestingly, the levels of anti-thrombin, fibrinogen, and Factor XIII levels were similar between CLTI and non-PAD controls. With the exception of anti-thrombin and Factor XIII, we noted a significant difference in all investigated proteins between the all PAD patients and non-PAD controls.
Discussion
Understanding the key players needed to initiate and regulate the coagulation cascade in patients with CLTI is essential in order to better manage them. Previous studies have shown that patients with CLTI are pro-thrombotic and have evidence of fibrinolysis relative to non-PAD controls. 7 This thrombotic state was also observed after arterial revascularization in patients with CLTI.30 The large Cardiovascular Outcomes for People Using Anticoagulation Strategies (COMPASS) trial also demonstrated several clinical benefits such as the reduction of cardiovascular death, stroke, myocardial infraction, and lower adverse limb events when PAD patients were treated with a combination of the low dose of the anticoagulant rivaroxaban and anti-platelet drug aspirin. 31 However, the reasons behind this finding are yet to be understood.
Despite several studies suggesting elevated levels of thrombin Frag1 + 2 in atherosclerotic disease, there are very few reports that have investigated thrombin activation (thrombin Frag 1 + 2 and TAT) in CLTI patients. 32 Relative to non-PAD controls, we demonstrated increased levels of thrombin frag 1–2, TAT, and D-dimer which are indicative of coagulation cascade and the fibrinolytic system activation in the CLTI group but not in PAD patients with claudication. In addition to coagulation cascade activation in CLTI group, we also noticed increased circulatory levels of markers indicative of platelet activation, namely PF4, P-selectin, and sVCAM-1. A similar finding was previously reported in patients with acute myocardial ischemia. 33 Interestingly, in the CLTI group, we noticed markers of platelet activation in patients despite being managed with anti-platelet medication. Although a larger study is needed to confirm our findings, we are proposing that relative to non-PAD patients, the observed pro-thrombotic state observed in all PAD patients is mainly driven by the CLTI patient population.
In addition, we investigated several biomarkers that are closely related to the activation of the coagulation cascade, namely inflammation and endothelial injury pathways. Relative to non-PAD controls, a common theme that we observed is the elevation of inflammatory cytokines and biomarkers of endothelial injury mainly in the CLTI cohort but not in patients with claudication. It is unclear if the observed increase in these pro-thrombotic proteins in CLTI patients is the cause of disease progression or is the effect of CLTI disease state. However, this demonstrates a strong association between these proteins and CLTI. There are some reports in other models of atherosclerosis which suggest a similar increase in circulating levels of markers indicative of endothelial dysfunction, namely TF, TM, and E-selectin.34,35 Such an increase in evidence of endothelial injury in CLTI may trigger the activation of the coagulation cascade and help explain the observed pro-thrombotic state in CLTI patients. Moreover, the enhanced inflammatory response in patients with severe atherosclerosis is well documented in the literature, which results in elevation of CRP, ADP, GDF-15, and L-selectin.36–39 Relative to non-PAD controls, we noted a trend of enhanced inflammatory response in CLTI patients and not in PAD patients with claudication. Some of these inflammatory proteins have been described as predictor of mortality.36,39 This finding parallels the documented high rate of mortality (25% at two years) observed in patients with CLTI patients. 40 However, it is essential to note that our data provide only a snapshot of the investigated interlinked pathways in PAD patients.
Finally, we wanted to understand some possible factors that can explain the findings that we observed. For the first time, we have demonstrated a significant reduction in circulating levels of the natural anti-coagulant protein C (over two-fold decrease) in CLTI patients relative to controls. Protein C and its co-factor protein S are involved in inhibiting the activity of activated FVIII and activated FIX. The loss of inhibitory effects of proteins C and S will promote a thrombogenic coagulation state. 41 Therefore, a reduction in proteins C and S may help explain why CLTI patients are more thrombogenic, with higher levels of TAT and Frag1-2. This reduction in protein C and S levels led us to study the concentration levels of coagulation factors in patients with PAD. As shown in our results section, CLTI patients had a significant reduction in the circulating levels of coagulation factors of the intrinsic system. Once again, it is not clear if the reduction in these clotting factors and regulators cause CLTI or the reduced levels are a consequence of CLTI. There are a few reasons that can explain our observation: the reduction in clotting factors might be secondary to the negative feedback loop induced by the activated thrombin or these factors are depleted secondary to their consumption in clot formation as a result of coagulation system activation. Two reports noted similar findings in patients with coronary arterial disease.42,43 These reports proposed a mechanism of a depleted state of coagulation factors in a thrombotic state secondary to atherosclerosis. So, the same theory can hold true for patients with CLTI.
This present study also has some limitations. Despite having a relatively small sample size for most of our experiments, a larger patient population would have been ideal to further strengthen our findings. Secondly, associating the biochemical findings with adverse clinical outcomes such as death, limb amputation, and cardiac events are helpful to better understand the correlation between coagulation status and study events. We are currently following up on our patients to investigate a potential clinical correlation. Finally, the biomarkers were measured only on the index admission or visit of the patients. Multiple measurements taken over duration of time may be more informative, especially in patients with deterioration in their clinical PAD status.
Altogether, in this paper, we demonstrated that patients with CLTI are hypercoagulable in relation to non-PAD controls. Our data also demonstrate that PAD patients with claudication had similar circulating levels of biomarkers indicative of a pro-thrombotic state as the control non-PAD cohort. We propose that the hypercoagulable state in CLTI is secondary to multiple factors including platelet activation, endothelial injury, loss of natural anti-coagulants, and pro-inflammatory state. Although larger studies are needed, our data favor the hypothesis that precision medicine in the form of aggressive medical optimization may be beneficial for patients with CLTI.
Footnotes
Acknowledgements
Some of the contents of this manuscript were presented at the 41st Annual Meeting on Vascular Surgery, hosted by The Canadian Society for Vascular Surgery on 13–14 September 2019 in Kelowna, British Columbia, Canada.
Declaration of conflicting interests
The author(s) declared following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Subodh Verma holds a Tier 1 Canada Research Chair in Cardiovascular Surgery and reports receiving research grants and/or speaking honoraria from Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly, EOCI Pharmacomm Ltd, Janssen, Merck, Novartis, Novo Nordisk, Sanofi, Sun Pharmaceuticals, and the Toronto Knowledge Translation Working Group. He is also the President of the Canadian Medical Surgical Knowledge Translation Research Group, a federally incorporated not-for-profit physician organization. All other coauthors declare that there is no conflict of interest.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by a grant from the Blair Foundation grant, as Dr. Qadura is a Blair Early Career Professor in Vascular Surgery at the University of Toronto.