
Abstract
Objectives
Cardiovascular disease (CVD) remains the primary cause of morbidity and mortality worldwide. The abnormal proliferation of vascular smooth muscle cells (VSMCs) is a key event in the pathogenesis of CVD. The functional and phenotypic changes in vascular cells are mediated by complex signaling cascades that initiate and control genetic reprogramming. Many studies have demonstrated that signal transducer and activator of transcription 3 (STAT3) regulates a diverse array of functions relevant to atherosclerosis.
Methods
In this review, we summarize the studies on the STAT3-mediated proliferation of VSMCs and subsequent CVDs such as hypertension, atherosclerosis, stroke, coronary artery disease, and myocardial infarction. Furthermore, we describe the general background of STAT3, its structure, function and regulation as well as the STAT3 signaling pathway. Finally, we highlight some potential issues and propose some solutions to these issues.
Introduction
Cardiovascular diseases (CVD), such as hypertension, atherosclerosis, stroke, coronary artery disease and myocardial infarction, continues to increase in prevalence. CVD remains the primary cause of morbidity and mortality, and still presents a significant burden worldwide. Globally, it has been estimated that there are 17.8 million deaths from CVD, which is nearly a third of total deaths in the year 2017 and increased by 21.1% between 2007 and 2017. 1 Many advances including medications and surgery in therapies of CVD have been made. However, various treatment options being only partially beneficial underscore the necessity of investigating CVD pathogenesis.
Vascular smooth muscle cells (VSMCs) are the main cellular components of the vascular system and play a critical role in the conductive function of the vasculature by maintaining the integrity of the vessel wall and adjusting arterial tone. 2 VSMCs maintain considerable plasticity and show varied phenotypes in response to environmental influences, allowing them to carry out many functions. 3 In response to environmental stimuli, VSMCs play an indispensable role in vascular repair by dramatically increasing the rate of proliferation, migration, and synthesis. 4 Many studies have demonstrated that VSMC proliferation is a vital factor in the development of multifarious CVDs. Phenotypic transformation is a major initiating factor of vascular remodeling and is attributed to the excessive proliferation of VSMCs, which are thought to influence the vascular pathological conditions of hypertension and atherosclerosis. 5 Moreover, the abnormal proliferation of VSMCs causes vascular remodeling that manifests as vessel wall thickening, which damages the smoothness of blood flow; consequently, blood pressure rises, ultimately causing CVD. 6
Transducer and activator of transcription 3 (STAT3) was first discovered in 1994, and its ability to promote cellular proliferation has gradually been uncovered. Previous studies have verified that this ability plays an essential role in various diseases, especially cancer. 7 Emerging studies have revealed that STAT3 is a crucial transcription factor that mediates signaling involved in cellular proliferation and differentiation, apoptosis, inflammation, and immunity, and these processes play a significant role in the initiation and continuation of various pathophysiological changes in CVD. 8 Cell cycle progression is important for cellular proliferation, and the cell cycle is divided into four phases: G0/G1, S, G2, and M. 9 In normal arteries, VSMCs are always quiescent and remain in the G0/G1 phase, but when VSMCs are irritated by vascular pathologies, the cyclin and cyclin-dependent kinase (CDK) complex forms, resulting in VSMCs exiting G1 phase and entering S phase.10,11 DNA replicates in the S phase, and in the next phase G2, extra mRNAs and proteins are prepared for mitosis, leading to VSMC proliferation. 12 In summary, abnormal increases or decreases in cell factors, growth-regulating factors, and vasoactive substances can accelerate VSMC proliferation under pathological conditions.
VSMC proliferation is of great significance in the progression of CVD. STAT3 can be regulated by a variety of signals and regulates other cellular regulatory factors, thereby participating in the proliferation of VSMCs. Thus, studying the mechanism of VSMC proliferation induced by the STAT3 pathway is valuable for finding therapeutic targets for CVD.
The structural and functional characteristics of STAT3
STAT protein transcription factors are latent in the cytoplasm and are essential for cytokine and growth factor signaling. 13 Mammalian STATs are composed of six structural regions, including the N-terminal domain (ND), coiled-coil domain (CCD), DNA-binding domain (DBD), linker domain (LD), SH2 domain, and transactivation domain (TAD). 14 Upon receptor activation, a single tyrosine residue is phosphorylated at the STAT3 705 site (Tyr705); then, STAT3 forms homodimers or heterodimers through reciprocal SH2-phosphotyrosine interactions, translocates to the nucleus, and promotes gene transcription (Figure 1). 15 STAT3 has two different isoforms, STAT3α (approximately 89–92 kDa) and STAT3β (approximately 80–84 kDa). STAT3β lacks the 55 C-terminal amino acids of STAT3α and has specific 7 amino acid residues. 16 STAT3α plays a key role in the modulation of cellular responses to interleukin-6 (IL-6) and in the mediation of interleukin-10 (IL-10); expressing STAT3β can rescue the embryonic lethality of a STAT3-null mutation and activate the expression of specific STAT3 target genes. 17 Additionally, the stability of activated dimer formation is key in determining the DNA-binding activity of STAT3 isoforms, and STAT3β dimers are more stable than STAT3α dimers because the acidic C-terminal region destabilizes STAT3α dimers. 18
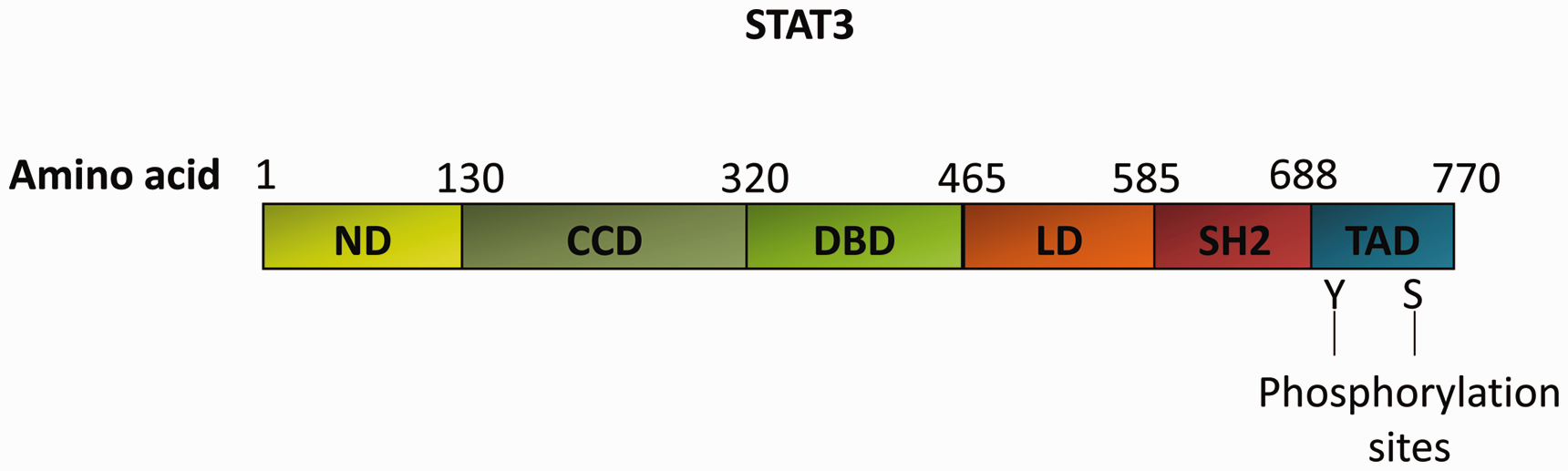
Structural characteristics of STAT3α. STAT3α includes six domains: a helical N-terminal domain (ND); a coiled-coil domain (CCD); a central DNA-binding domain (DBD); a linker domain (LD); a Src homology 2 domain (SH2); and a C-terminal transactivation domain (TAD) with a conserved tyrosine residue at 705 (Y705) and a serine phosphorylation site at 727 (S727).
Unphosphorylated STAT3 (u-STAT3) induces cytokine-dependent signaling through a mechanism entirely different from that used by phosphorylated STAT3 (p-STAT3). The expression of u-STAT3, which depends on signal enhancement, is important for regulating gene expression and physiological responses to cytokines and growth factors that activate STAT3. 19 One distinguishing characteristic of STAT3 is its capacity to activate different sets of genes in various cells to affect cell growth, proliferation, and apoptosis. 20 Genes that regulate cell survival include bcl-2, bcl-xL, and Fas, 21 and genes encoding cell cycle regulatory proteins, including cyclin D1, D2, D3, p21, and p27, are direct targets of STAT3. 22
Notably, multiple signaling pathways involving STAT3 are involved in the proliferation of VSMCs (Figure 2), and STAT3-dependent regulation appears to be complex and diverse. STAT3 protein plays a critical role in VSMC proliferation as a significant signaling molecule.
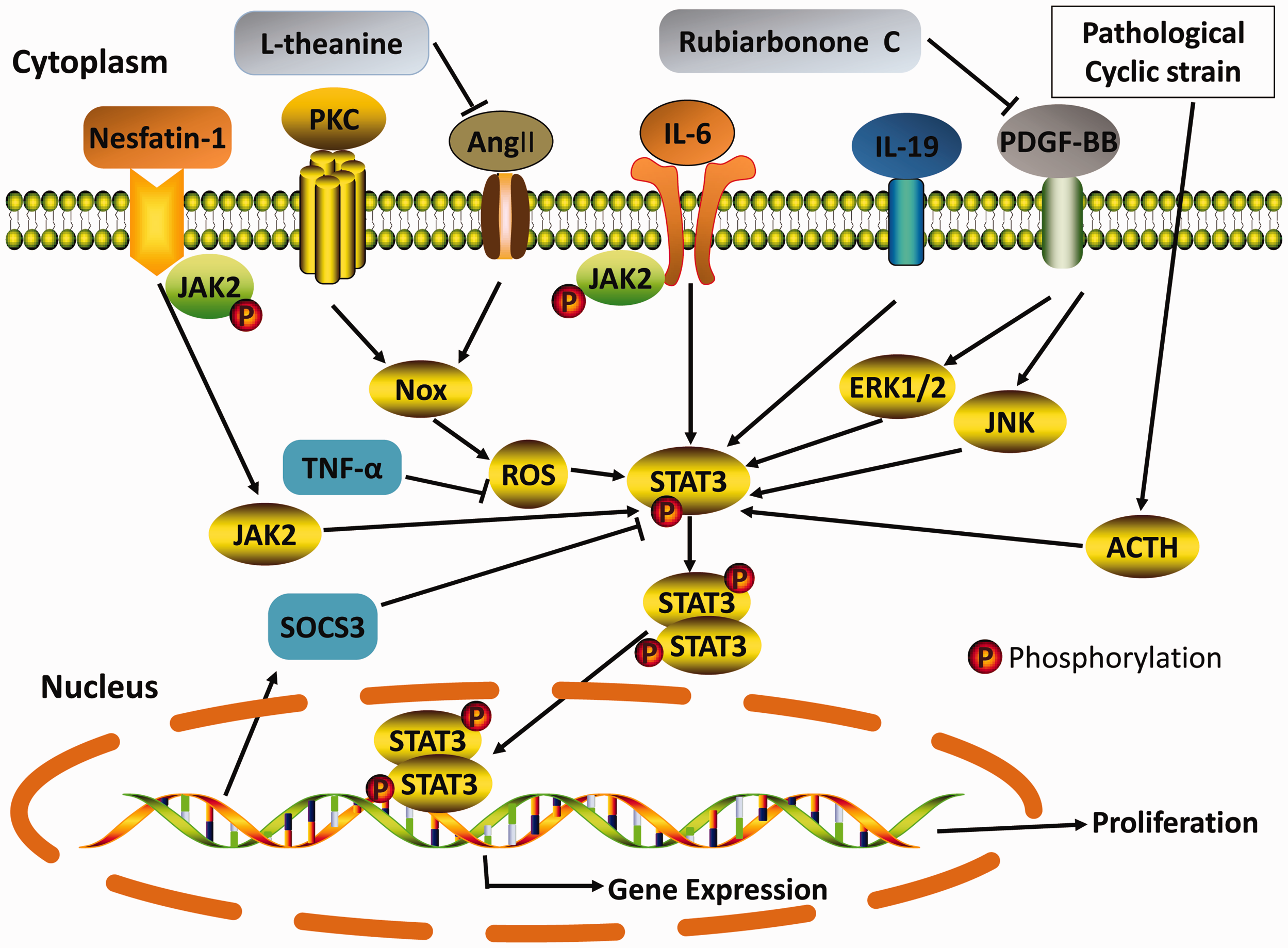
Physiological regulation and pharmacological inhibition of the STAT3 signaling pathway. STAT3 is an essential player in cell signaling and transcription in response to a wide range of stimuli and acts via various mechanisms. The most relevant STAT3 signaling pathways in VSMC proliferation are indicated with continuous lines. The whole process involves seven classes of STAT3-associated pathways. The combination of these mechanisms leads to the proliferation of VSMCs. Nesfatin-1; PKC: protein kinase C; Ang II: angiotensin II; IL-6: interleukin-6; IL-19: interleukin-19; PDGF-BB: platelet-derived growth factor-BB; ACTH: adrenocorticotropic hormone; JAK2: Janus kinase 2; Nox: nicotinamide adenine dinucleotide phosphate oxidase; ROS: reactive oxygen species; ERK1/2: extracellular signal-regulated kinase 1/2; JNK: c-Jun N-terminal kinase; STAT3: signal transducer and activator of transcription 3; TNF-α: tumor necrosis factor α; SOCS3: suppressor of cytokine signaling 3; L-theanine; Rubiarbonone C.
STAT3 signaling pathway is involved in VSMC proliferation
Nox/ROS/STAT3 signaling pathway
Reactive oxygen species (ROS) are principally produced by the mitochondria in normal cells, and the three major types of ROS are hydrogen peroxide (H2O2), hydroxyl radicals (OH), and superoxide anion radicals (O2¯). 23 ROS are closely associated with vascular cell proliferation, migration, and hypertrophy. 24 Moreover, ROS formed in the vascular wall target a wide range of signaling molecules and cellular pathways in vascular smooth muscle, 25 including the Janus kinase (JAK)/STAT3 pathway. 26 The JAK/STAT3 pathway is activated in diverse cellular processes and plays an essential role in the intracellular mechanism of tumor necrosis factor α (TNF-α) and other stimuli regulating gene expression; 27 TNF-α can inhibit the activation of redox-sensitive protein kinases and transcription factors in VSMCs. 28 ROS contribute to the pathogenesis of CVD; in atherosclerotic lesions, TNF-α is secreted by VSMCs in the neointima after balloon injury, and macrophage stimulation is a common factor in ROS production. 29
Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-derived ROS production is involved in the growth, apoptosis, and migration of VSMCs. 30 In addition, NADPH oxidase (Nox) catalyzes molecular oxygen reduction to form superoxide, resulting in the proliferation of VSMCs and increasing the transduction of proinflammatory signals in VSMCs. 31
Ang II/Nox/ROS/STAT3
Angiotensin II (Ang II) produced by angiotensin-converting enzyme (ACE) comprises the classical renin-angiotensin system (RAS), which regulates and maintains vascular tone, blood pressure, and fluid balance. 32 Responsiveness to Ang II is mainly conferred by the expression of angiotensin receptor 1 (AT1). Prior studies have shown that Ang II induces Nox production, leading to increased ROS production and eventually regulating VSMC proliferation. 33 Ang II stimulates upstream c-Src tyrosine kinase, and JAK and STATs are impacted downstream. 34 Evidence demonstrates that atrial structural remodeling is mediated by the Ang II/STAT3 pathway. 35 In the atrium, Ang II can activate STAT3 and induce apoptosis, collagen production, and matrix metalloproteinase expression; finally, Ang II may induce structural changes in the atrium. 36
Abnormal VSMC proliferation is the structural foundation of hypertension. 37 Moreover, hyperglycemia increases Ang-II-induced VSMC proliferation by increasing JAK2/STAT3 pathway transduction. 38 In contrast, partly by obstructing the JAK/STAT3 pathway, L-theanine restrains the Ang II-induced proliferation and migration of VSMCs. 39 Others have reported that Angs 1–7 protect against injury and inflammation in human umbilical vein endothelial cells (HUVECs) by inhibiting the activation of the JAK2/STAT3 pathway. 40
PKC/Nox/ROS/STAT3
The protein kinase C (PKC) family is a group of enzymes that depend on calcium and phospholipids and play an important role in cellular signaling systems. 41 PKC has been shown to regulate various signal transduction events implicated in the pathogenesis of inflammation, such as inflammatory cytokine and superoxide biosynthesis and phospholipase A2 (PLA2) activity. 42 PKC plays a pivotal role in mediating ROS generation and regulating the activation of Nox, 43 which is dependent on high glucose-stimulated aortic endothelial cells, smooth muscle cells, and renal mesangial cells. 44 Adenosine 5′-O-(3-thiotriphosphate) (ATPγS) stimulates the activation and translocation of STAT3 via the PKC-dependent ROS generation pathway in VSMCs, and this induces cytosolic phospholipase A2 (cPLA2)/cyclooxygenase-2 (COX-2)/prostaglandin E2 (PGE-2) signaling, ultimately leading to the proliferation of VSMCs. 45 Moreover, toll-like receptors (TLRs) inhibit the expression of regulator of G-protein signaling 2 (RGS2) through the PKC-η/phospholipase D2 (PLD2) pathway, eventually upregulating Nox1 expression by dissociating STAT3 from its repressor RGS2. 46
Overall, the Nox-induced, ROS-stimulated activation of STAT3 increases the proliferation of VSMCs, which is a key pathway for VSMC proliferation. Ang II and PKC can regulate ROS activity by increasing Nox. Notably, hyperglycemia enhances angiotensin Ang-II-induced VSMC proliferation by activating JAK2/STAT3 pathway signal transduction.
PDGF-BB/MAPK/STAT3 signaling pathway
Platelet-derived growth factor (PDGF)-BB is primarily released by platelets at injured vascular sites, and it regulates transcription factors and critical molecular signaling pathways, leading to the promotion of VSMC proliferation and migration.47,48 The role of PDGF in enhancing VSMC proliferation is well recognized. Furthermore, research has shown that STAT3 is specifically required for the PDGF-induced proliferation of human arterial smooth muscle cells (HASMCs) and the regulation of the cell cycle regulators cyclin D3 and p27. 49 Rubiarbonone C, one of the major triterpenoids of the Rubia philippinensis Elmer (family Rubiaceae) plant, can promote PDGF-BB-induced cellular proliferation and migration by inhibiting the extracellular signal-regulated kinase 1/2 (ERK1/2) MAPK and c-Jun N-terminal kinase (JNK) signaling pathways, which specifically control STAT3 activation. 50
In addition, many miRNAs have been shown to play a regulatory role in VSMC proliferation and migration. The overexpression of miR-137 inhibits VSMC proliferation and migration; moreover, PDGF-BB treatment increases the expression of miR-137 in VSMCs. 51 MiR-145 also inhibits VSMC proliferation by regulating CD40. 52 However, miR-503 suppresses PDGF-BB-induced human aortic VSMC proliferation and migration by targeting the insulin receptor. 53 MiR-34a activates the proliferation of human pulmonary artery smooth muscle cells (HPASMCs) by targeting PDGF-α. 54 MiR-181b activates the PI3K and MAPK signaling pathways and subsequently enhances VSMC proliferation. 55
IL/STAT3 signaling pathway
IL-6/JAK2/STAT3
IL-6 is an archetypal cytokine that plays a significant role in hematopoiesis, tissue homeostasis, metabolism, and immunity. IL-6 activates JAK2/STAT3, inducing VSMC proliferation. 56 IL-6-induced VSMC motility relies on the phosphorylation of glycoprotein 130 (gp130)/STAT3 signaling and induces cyclin D1 expression. 57 In contrast, epidermal growth factor (EGF) is also involved in VSMC proliferation. 58 Heparin-binding EGF-like growth factor (HB-EGF) delays STAT3 phosphorylation via NF-κB expression and IL-6 secretion in VSMCs. 59 Research suggests that IL-6 plays a key role in the pathogenesis of atherosclerosis and coronary restenosis. 60 Overall, IL-6 induced by HB-EGF plays an important role in delaying STAT3 activation and VSMC proliferation.
IL-19/STAT3
IL-19 is a member of the IL-10 family and plays an important role in immunological diseases. IL-19 has protective effects on VSMCs, prominently decreasing neointimal hyperplasia and the proliferation of VSMCs in balloon angioplasty-injured rat carotid arteries. 61 In addition, IL-19 promptly activates STAT3, indicating that IL-19/STAT3 mediating suppressor of cytokine signaling 5 (SOCS5) expression is a likely mechanism by which IL-19 might exert its anti-proliferative and anti-restenotic effects. 62 SOCS3 overexpression has been found to observably depress VSMC proliferation and neointimal hyperplasia. 63 However, STATs are essential for IL-19-driven oxygenase-1 (HO-1) expression, 64 and HO-1 protects against vascular inflammation by decreasing monocyte arterial transmigration induced by oxidized low-density lipoprotein (oxLDL), decreasing VSMC proliferation, and acting as a potent antioxidant. 65
IL-19 has been shown to be involved in coronary artery disease and atherosclerosis. Thus, phosphorylated STAT3 forms a dimer in the nucleus and regulates the expression of genes, including NF-κB, cyclin D1, and SOCS3. Interleukin-induced STAT3 activation is also closely connected with VSMC proliferation.
Other STAT3 signaling pathways
Nesfatin-1/JAK2/STAT3
Nesfatin-1 is an anorexigenic peptide from nucleobindin 2 (NUCB2), 66 and it modulates many important processes, such as glucose homeostasis, lipid metabolism, and cardiovascular effects. Nesfatin-1-induced activation of the interaction between PI3K/Akt/mTOR signaling and JAK2/STAT3 signaling promotes VSMC differentiation and proliferation and contributes to hypertension. 67
ACTH/ERK/STAT3
When VSMCs are exposed to high levels of cyclical strain, VSMCs undergo abnormal proliferation, leading to vascular remodeling 68 and increasing the expression of adrenocorticotropic hormone (ACTH) and melanocortin receptor type 2 (MC2R). 69 In addition, ACTH-dependent cell proliferation is closely associated with the phosphorylation of extracellular ERKs and STAT3. 69
CD36/STAT3
CD36 is a multifunctional receptor that reduces the expression of the antioxidant transcription factor NF-E2-related factor-2 (Nrf2) and subsequently downregulates the expression of specific antioxidant enzymes, including peroxiredoxin 2 (Prdx2) and heme oxygenase-1 (HO-1) in VSMCs. 70 Both Prdx2 and HO-1 have been reported to inhibit VSMC growth. 71 CD36 promotes VSMC proliferation via the upregulation of cyclin A expression and increases dedifferentiated VSMC phenotypes via the inhibition of STAT 3 activity. 72
Physiological roles of other STATs in VSMC proliferation
The STAT family comprises seven known members: STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6. The ability of STATs to regulate cellular proliferation has also been well documented; in addition, various cytokines can activate one type of STAT. In VSMCs, the expression of ZO-2 is upregulated in response to vascular injury; this change inhibits STAT1 expression and leads to accelerated cell growth and reduced vascular remodeling. 73 There have been few reports about the role STAT4 plays in VSMCs, and it has a potent effect on regulators of VSMC proliferation. 74 In contrast, STAT4 protein expression in VSMCs is associated with SOCS-3 and MCP-1 production. 75 IL-5 notably downregulates the fundamental process of VEGF-induced human lung vascular endothelial cell proliferation. Notably, STAT5 is critical for this effect of IL-5. 76 STAT6 signaling may contribute to the gene expression modifications underlying VSMC activation in the context of hyperplastic lesion formation, 77 and IL-4 induces the proliferation of STAT6-deficient lymphocytes. 78 Moreover, STAT6 may play a “permissive” role in cellular proliferation by downregulating the expression of p27 Kip1, a major inhibitor of cell cycle progression. 79
Conclusions and perspectives
VSMC proliferation plays a key role in the development of multifarious CVDs; the abnormal proliferation of VSMCs causes an increase in vessel wall thickness, which damages the smoothness of blood flow and increases blood pressure. In particular, under pathological conditions, abnormal increases or decreases in the levels of various cytokines can stimulate VSMC proliferation. STAT3 can be regulated by a variety of signals, including ROS, Ang II, PKC, PDGF, IL-6, IL-19, ACTH, and nesfatin-1, and STAT3 participates in the proliferation of VSMCs. Over the last two decades, STAT3 signaling has been identified as a pivotal pathway for the stimulation of vascular cells, especially under pathological conditions. A large number of studies have been published that describe the role of STAT3 signaling and VSMCs in CVD. There are a small number of articles about STAT3 signaling-induced VSMC proliferation, but the number of reports about this subject has gradually increased over the last 10 years. It is important to study the mechanism by which STAT3 induces VSMC proliferation and to find therapeutic targets for heart disease. Finally, the function of STAT3 in VSMC proliferation is an area that requires further investigation, and ultimately, researchers will find new therapeutic approaches to treat CVD.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (81202535), the Hunan Provincial Department of Education (nos. 19K084 and 19C1722), the Hunan Provincial Science and Technology Department (2018JJ6097), the Science and Technology Bureau of Chenzhou, Hunan Province (zdyf201925), and the Research Center of Diagnosis and Treatment Technology for Lipid Metabolism Disorders of Chenzhou, Hunan Province (yfzx201907).