
Abstract
ICH Q3D defines a science and risk-based assessment process to identify, evaluate, and define controls to limit elemental impurities in drug products. The present article comprise challenges and simplified approach to draw elemental impurities risk assessment and control in final drug product to satisfy current regulatory requirements using different calculation options. Detail risk assessment depends on information/knowledge of most significant potential sources of elemental impurities, i.e. drug product components, manufacturing process of drug substance or drug product, container closer system, etc. If there is lack of information available, the applicant may carry out end product testing as the initial strategy for risk assessment using option 3 calculation.
Keywords
Introduction
Control of metal impurities was primarily based on pharmacopoeial requirements for heavy metals, which was widely used for routine screening of pharmaceutical ingredients since the early 20th century. The commonly used methodology was mainly intended to control metals which form a sulphide precipitate, such as lead, copper and other metals. 1 ICH Q3D guidance comprises safety-based permitted daily exposures for 24 elementals and recommendations for a risk-based and science-based approach to control of those elements in pharmaceutical products. 2 New NDA and ANDA applications submitted after 1 June 2016 should follow the recommendations of Q3D. For existing marketed products, manufactures should follow the recommendations of Q3D and/or comply with USP <232> by 1 January 2018. 3
Challenges for the drug product manufacturer (applicant) regarding the health authority expectations on elemental impurities risk assessment submission 4
– Where to start assessing the elemental impurities risk? – Up to what level is the presentation of the risk assessment? – How much data are required to support risk assessment conclusions? – What are the most significant potential sources of elemental impurities? – Where the information should be presented in the dossier. – Category of supplement filing for products which are currently approved. – Periodic testing of elemental impurities in drug products and components of drug products.
Q3D Guideline clarifies the risk assessment to be considered for elemental impurities based on their toxicity and potential source (Fig. 1) in new drug applications (Table 1) and abbreviated new drug applications.
Potential source of elemental impurities.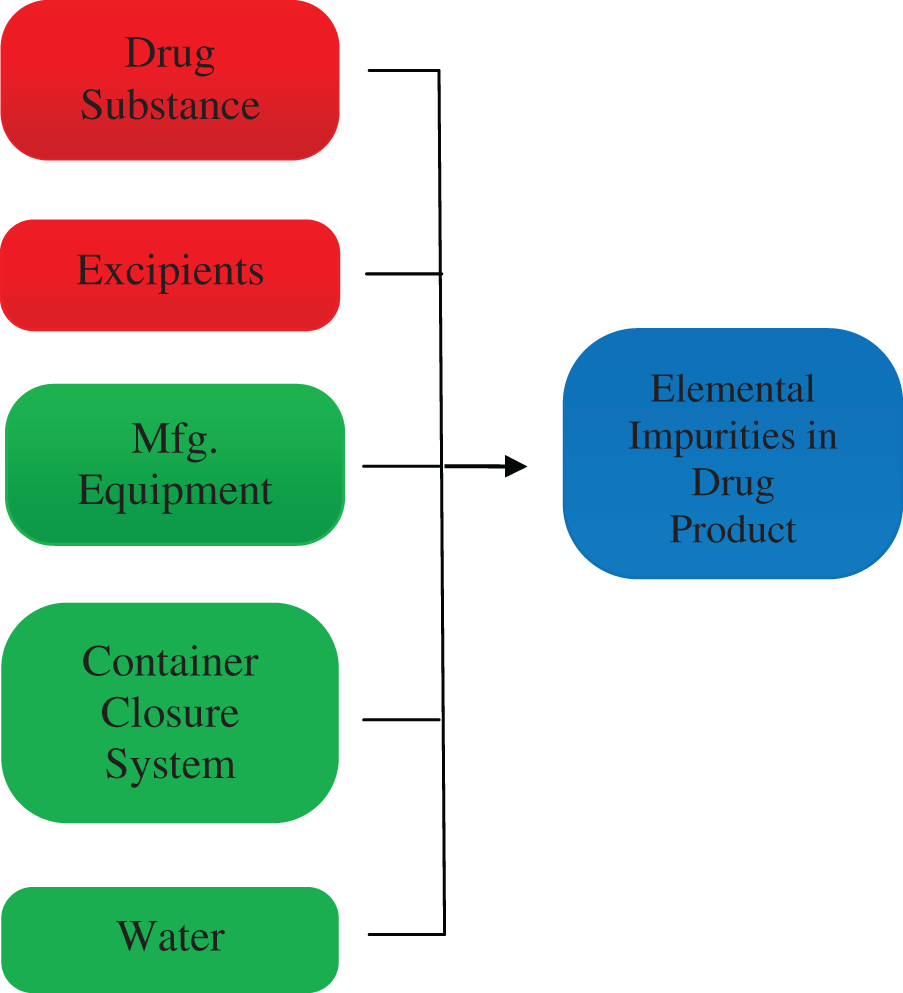
Guidance has provided PDE and the calculation options (option 1, option 2a & 2b, option3) to describe some acceptable approaches to establishing concentrations of elemental impurities in drug products or components that would assure that the drug product does not exceed the PDEs.
Different classes of elements based on toxicity and requirement to consider under risk assessment in finish drug product.

Practical approach to present risk assessment of Solid oral dosage form in regulatory submission
It is better to capture detailed elemental risk assessment of drug product in pharmaceutical development section (3.2.P.2) and summary of the risk assessment in justification of drug product specification (3.2.P.5.6) of regulatory dossier.
Case study 1
Composition of X tablet
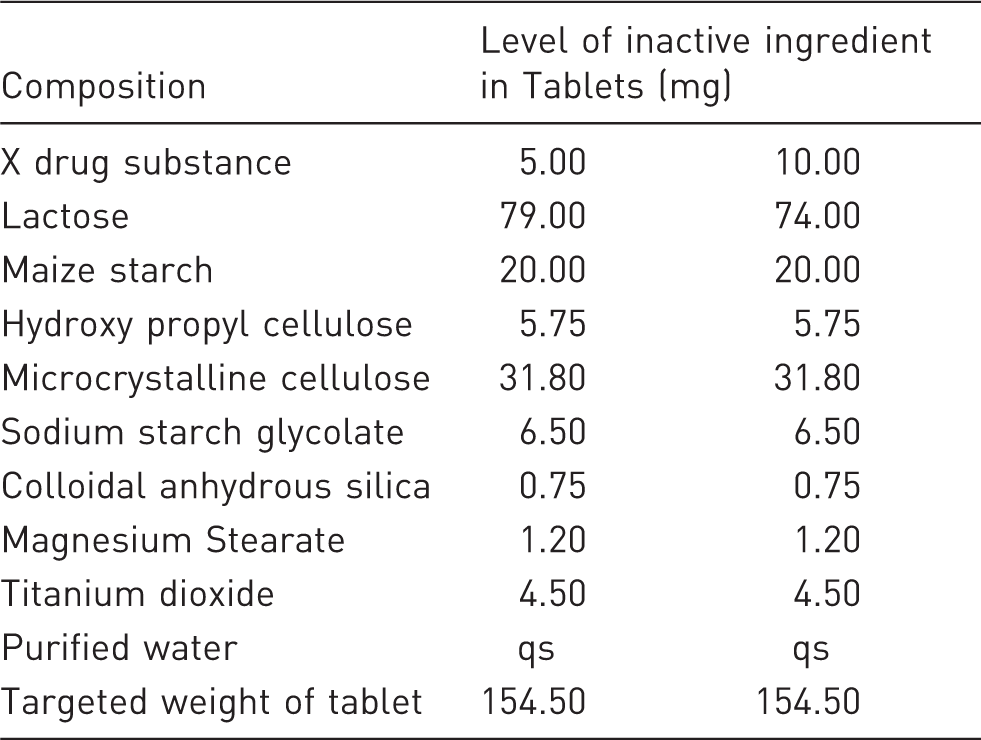
Container closer system: HDPE bottle with CR closer.
Supplier of each drug product component has provided limited information on elemental impurities present in each drug components, i.e.
– three of suppliers have provided statement that excipients provided are in compliance with ICH Q3(D) option 1. – two of suppliers have stated that provided drug substance or excipients are having possible elemental impurities with limit of below control threshold as per ICH Q3(D). – remained suppliers have provided observed level of possible elemental impurities with specification control limits. – all of suppliers of drug product component have also stated that there are no any elements intentionally added or used during synthesis of excipient.
With above information/knowledge provided regarding elemental impurities present in each drug product components, initial risk assessment can be drawn as mentioned in Table 3.
Initial evaluation of the elements to be considered in the risk assessment 6
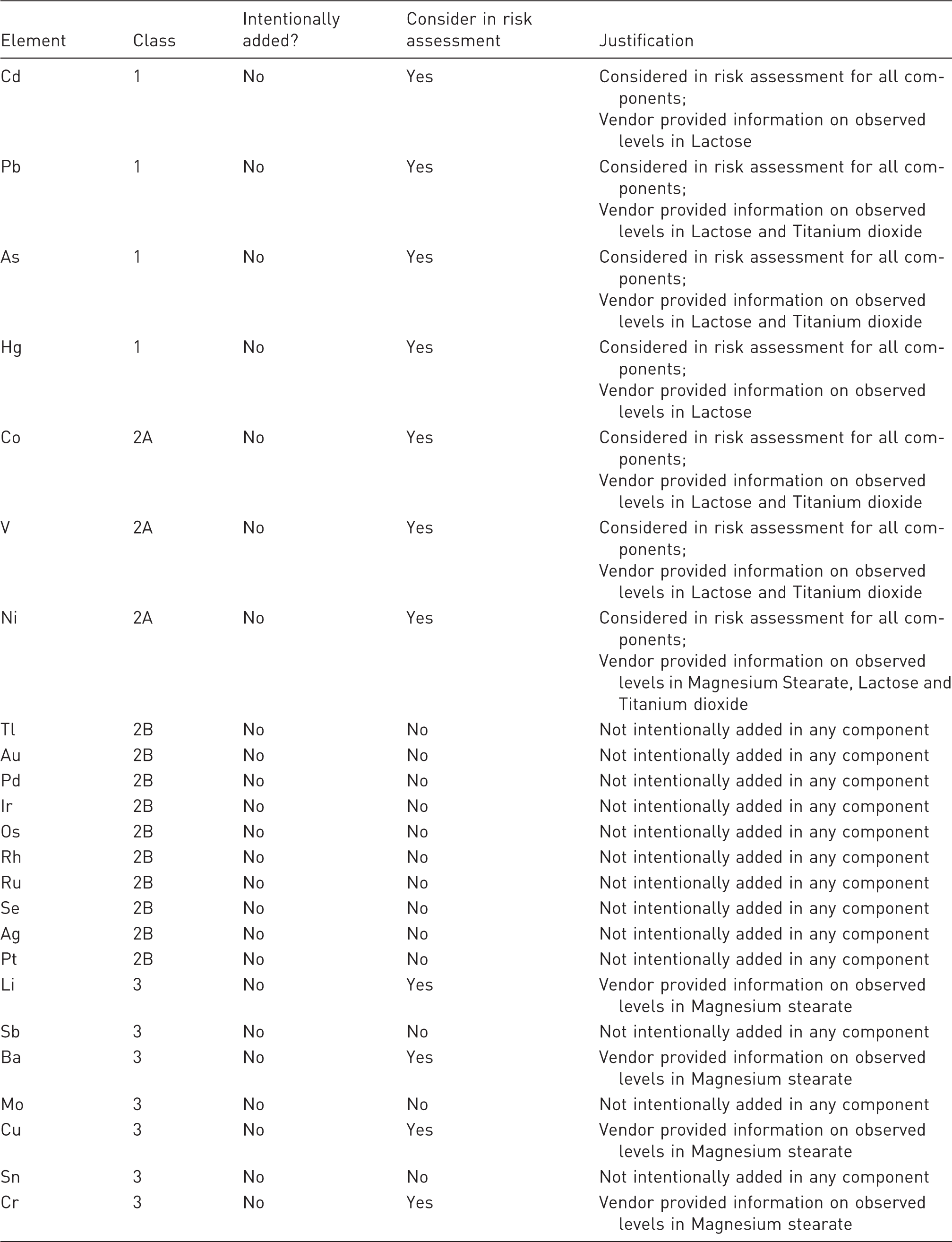
Evaluation of potential contributors of elemental impurities to the drug product

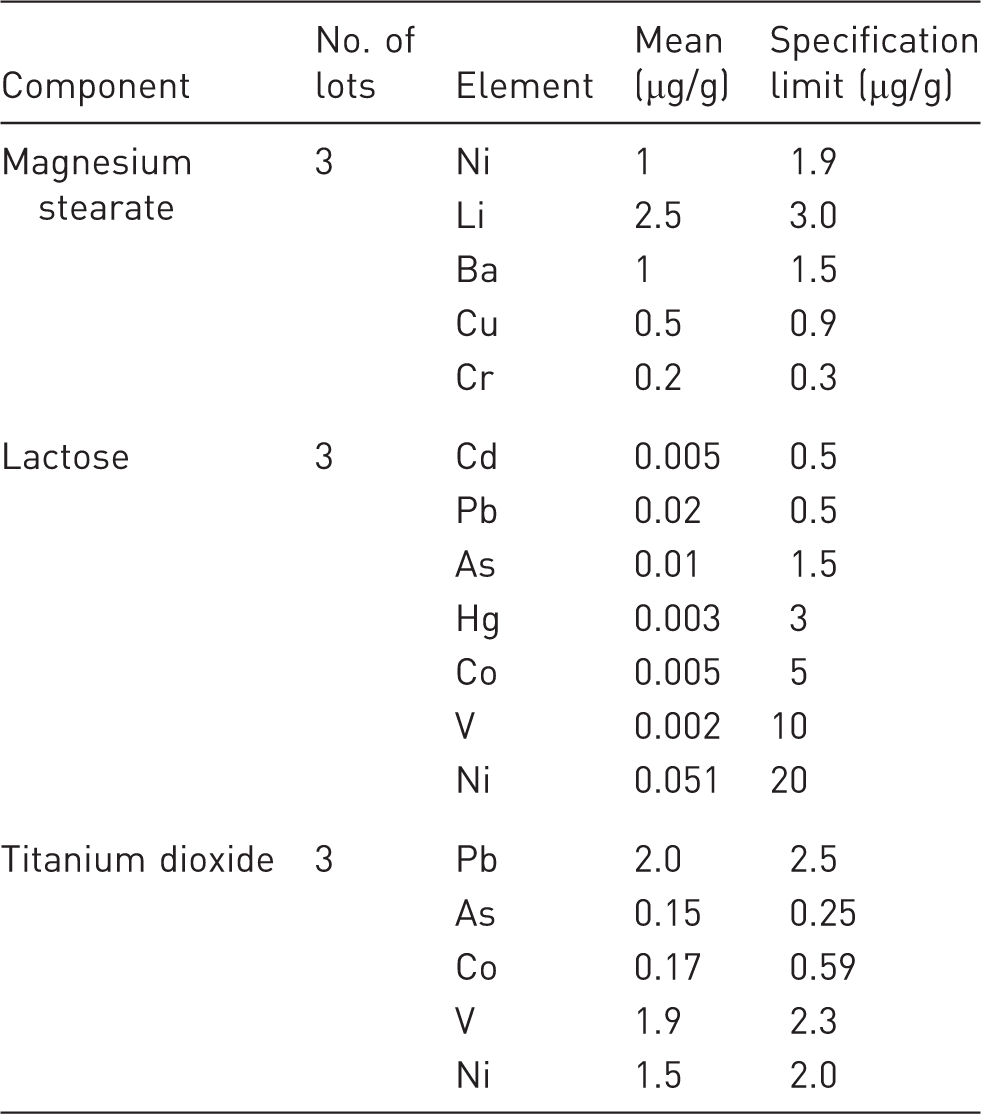
Analytical measurements were performed on three components by vendor to support the risk assessment. Elemental impurity data are generated using a validated method (see the Table 5).
Summary of elemental impurity data for potential components of X tablets
Calculated concentrations of elemental impurities in X tablets using established specification limits and Q3D Option 2B [for Case 1]

Maximum daily dose: 0.010 g per day. The total drug product administered would be 0.309 g (considering two tablets of 5 mg).
Conclusion (Case 1)
The risk assessment identified elemental impurities (i.e. Cd, Pb, As, Hg, Co, V, Ni, Li, Ba, Cu, Cr) that could be observed in three of the components of X tablets. The three components and the potential elemental impurity in each are: Magnesium Stearate (Ni, Li, Ba, Cu, Cr), Lactose (Cd, Pb, As, Hg, Co, V, Ni), and Titanium dioxide (Pb, As, Co, V, Ni).
An elemental impurity specification and associated limit are not proposed for X tablets; however, to ensure that identified elemental impurities levels are maintained at or below the respective PDE, a specification and limit for identified elemental impurities in three of the components were established. These incoming material controls ensure that the elemental impurities level will be maintained at or below the PDE in X tablets.
The risk assessment will be updated (as necessary) if the process is modified or suppliers of the drug product components are changed and that any subsequent changes to the control strategy will be reported to the Health Authorities within the current regulatory framework.
Case study 2
In the above-mentioned case study 1, specification limit of all target elements identified in the risk assessment is below the permitted concentration limit (PDE) except one, i.e. limit of Pb (2.5 µg/g) in Titanium dioxide.
Calculated concentrations of elemental impurities in X tablets using the permitted concentration limits and Q3D Option 1 [for Case 2]
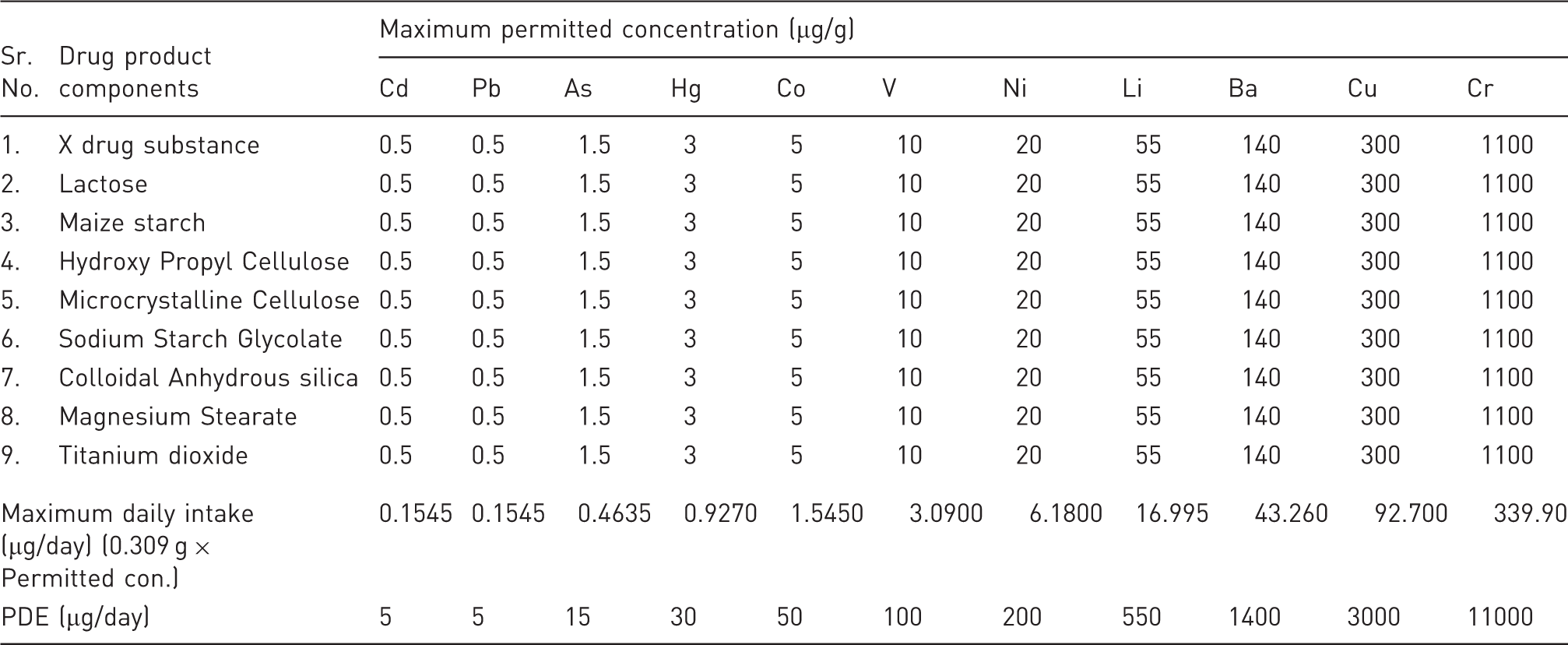
Conclusion (Case 2)
An elemental impurity specification and associated limit are not proposed for X tablets, because the identified elemental impurities levels do not exceed the option 1 concentration in all drug components and all these components can be used in any proportion in the X tablets.
The risk assessment will be updated (as necessary) if the process is modified or suppliers of the drug product components are changed and that any subsequent changes to the control strategy will be reported to the Health Authorities within the current regulatory framework.
In the above two cases, risk assessment has been drawn using information provided from drug component suppliers, knowledge of drug product manufacturing process and container closer system. Most of the drug product applicants may try to conclude elemental risk assessment using option 1 or 2 calculations in which final drug product analysis may not be required and it is a cost-effective approach.
Case study 3
Calculated concentrations of elemental impurities in X tablets using the Q3D Option 3 [for Case 3]
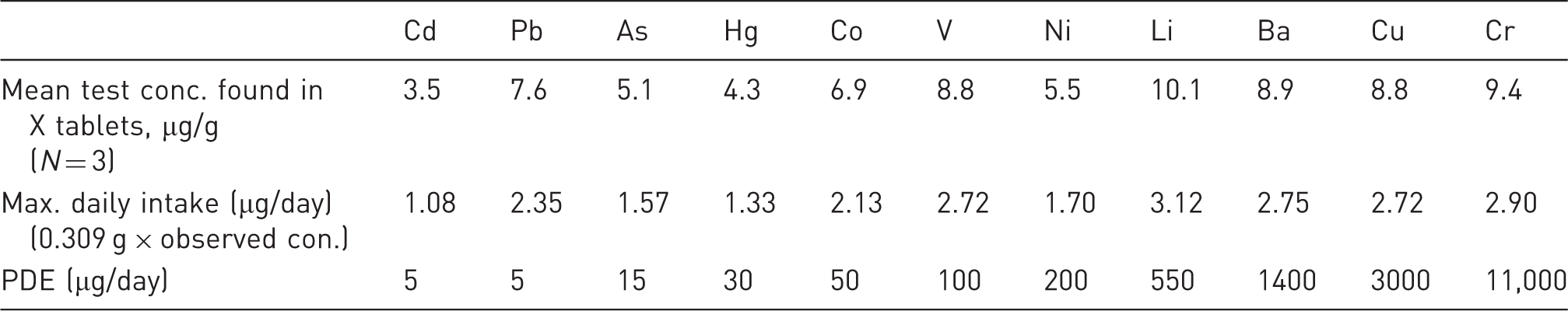
Elements of class 1, class 2a and other elements if identified in initial risk assessment must be covered into analysis of final drug product.
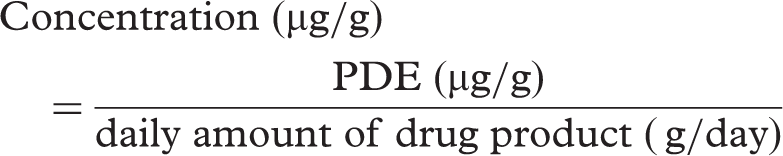
Maximum permitted concentrations of elemental impurities in X tablets using the daily intake of drug product and the PDE [for Case 3]
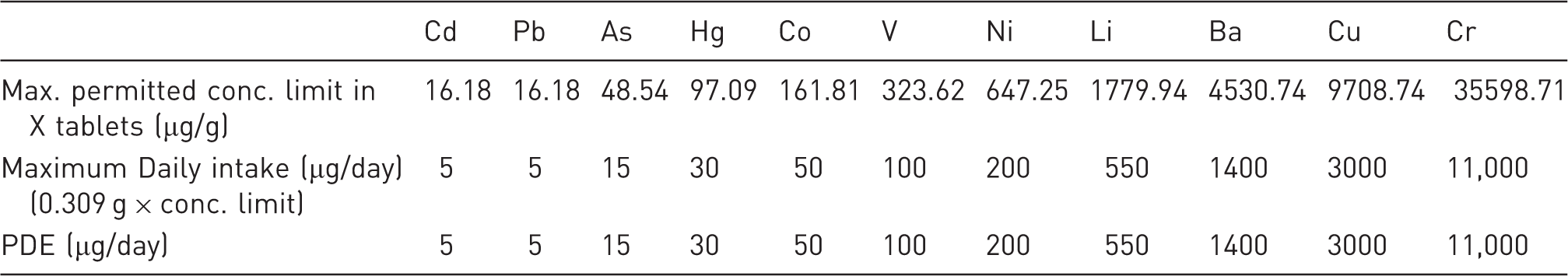
Conclusion (Case 3)
The observed elemental impurities levels in three lots of drug products are far below the permitted concentration limit in X tablets based on daily intake of drug product and PDE. Hence, an elemental impurity specification and associated limit are not proposed for X tablets.
The risk assessment will be updated (as necessary) if the process is modified or suppliers of the drug product components are changed and that any subsequent changes to the control strategy will be reported to the Health Authorities within the current regulatory framework.
Periodic Testing 8
Periodic testing may be applied to elemental impurities according to the principles described in ICH Q6A. Where the risk assessment indicates that routine testing is considered unnecessary but some additional assurance is needed post approval, and periodic testing of the drug product or one or more individual components may be proposed by the applicant and implemented upon acceptance by the regulatory authority. 8
Life-cycle approach to control strategy 8
Product and/or process changes across the product lifecycle have the potential to impact (positively or negatively) the elemental impurity content of the drug product. Changes can include, but are not limited to changes in synthetic routes, excipient suppliers, materials, processes, equipment, container closure systems or facilities. The impact of such changes on the original risk assessment should be evaluated, and implications for the control strategy should be considered. The regulatory implications of modifications to the risk assessment and control strategy should be considered, and appropriate variations should be submitted according to regional requirements. Consideration should be given for periodic review of the control strategy, as part of ongoing continual improvement. 8
Footnotes
Declaration of conflicting interests
The information is built from work, literature search and experience, and reflects the point of view of the author. It is not the official position of organizations identified above.
Funding
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The author(s) received no financial support for the research, authorship, and/or publication of this article.