
Abstract
The prevalence of the common LCHAD c.1528G>C p.(Glu510Gln) pathogenic variant in the HADHA gene in women and neonates with acute fatty liver of pregnancy is unclear. A review of nine case series comprising 113 pregnancies complicated by acute fatty liver of pregnancy, where maternal and/or fetal genetic testing was performed, found the LCHAD c.1528G>C p.(Glu510Gln) variant in only five cases (4.4%). In addition, an online survey disclosed no maternal and/or fetal pathogenic variants in 23 women who recalled the results of gene testing. Only a small minority of women with a clinical diagnosis of acute fatty liver of pregnancy have the common LCHAD c.1528G>C p.(Glu510Gln) variant on genetic testing.
Keywords
Introduction
Acute fatty liver of pregnancy (AFLP), estimated to complicate 1:7 000 to 1:20 000 pregnancies, is associated with significant maternal and fetal morbidity and mortality. Defects in fatty acid metabolism appear to play a role in pathogenesis, and several gene mutations leading to an abnormal fatty acid oxidation disorder (FAOD) have been associated with the development of AFLP, the most common of these being the homozygous LCHAD c.1528G>C p.(Glu510Gln) variant in HADHA associated with fetal long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHAD). Sequencing of DNA from 26 unrelated patients with LCHAD deficiency found the LCHAD c.1528G>C p.(Glu510Gln) pathogenic variant to be the only abnormality expressed in 24 individuals (92%), the remaining two patients being heterozygous for this mutation. 1 AFLP has been described with other FAODs, including short-chain acyl-CoA dehydrogenase (SCAD—three cases), carnitine palmitoyltransferase I deficiency (CPT1—two cases), medium-chain acyl-CoA dehydrogenase deficiency (MCAD—two cases), and single cases with long-chain 3-ketoacyl CoA thiolase enzyme and mitochondrial trifunctional protein deficiency (MTP), respectively.2–5 A case series of three Japanese women with AFLP/hemolysis elevated liver enzyme syndrome found that none of the women demonstrated the common LCHAD c.1528G>C p.(Glu510Gln) pathogenic variant, though all manifested homozygous wild-type genotype (c1/c1) in the 5’-flanking lesion of microsomal cytochrome P4502E1 (CYP2E1). 6 The prevalence of the LCHAD c.1528G>C p.(Glu510Gln) pathogenic variant in women diagnosed with AFLP is unclear. Estimates based upon individual case reports may be inaccurate due to reporting and publication bias.
Aim
The primary objective was to ascertain the prevalence of the LCHAD c.1528G>C p.(Glu510Gln) pathogenic variant in women or their offspring following a clinical diagnosis of AFLP in published case series. The common LCHAD c.1528G>C p.(Glu510Gln) mutation was the only variant tested for in seven of the nine case series where genetic testing was performed. Secondary objectives were to review the prevalence of pathogenic variants in published case reports and to review the literature regarding the prevalence of liver disease complicating pregnancies where the children were subsequently diagnosed with a FAOD.
Methods
Electronic databases (Ovid Medline, Ovid EMBASE, Google Scholar) were searched using the terms “acute fatty liver of pregnancy,” “LCHAD,” and “HADHA” (Figure 1). The search was conducted without language restriction from inception to May 2024. Two reviewers (AM, JH) assessed all titles and abstracts for eligibility. Abstracts were used to filter results and exclude duplicates. Inclusion criteria were case series and case reports of AFLP, and case series examining maternal liver disease in pregnancies where the fetus was subsequently identified with a FAOD. Studies with insufficient clinical information were excluded.
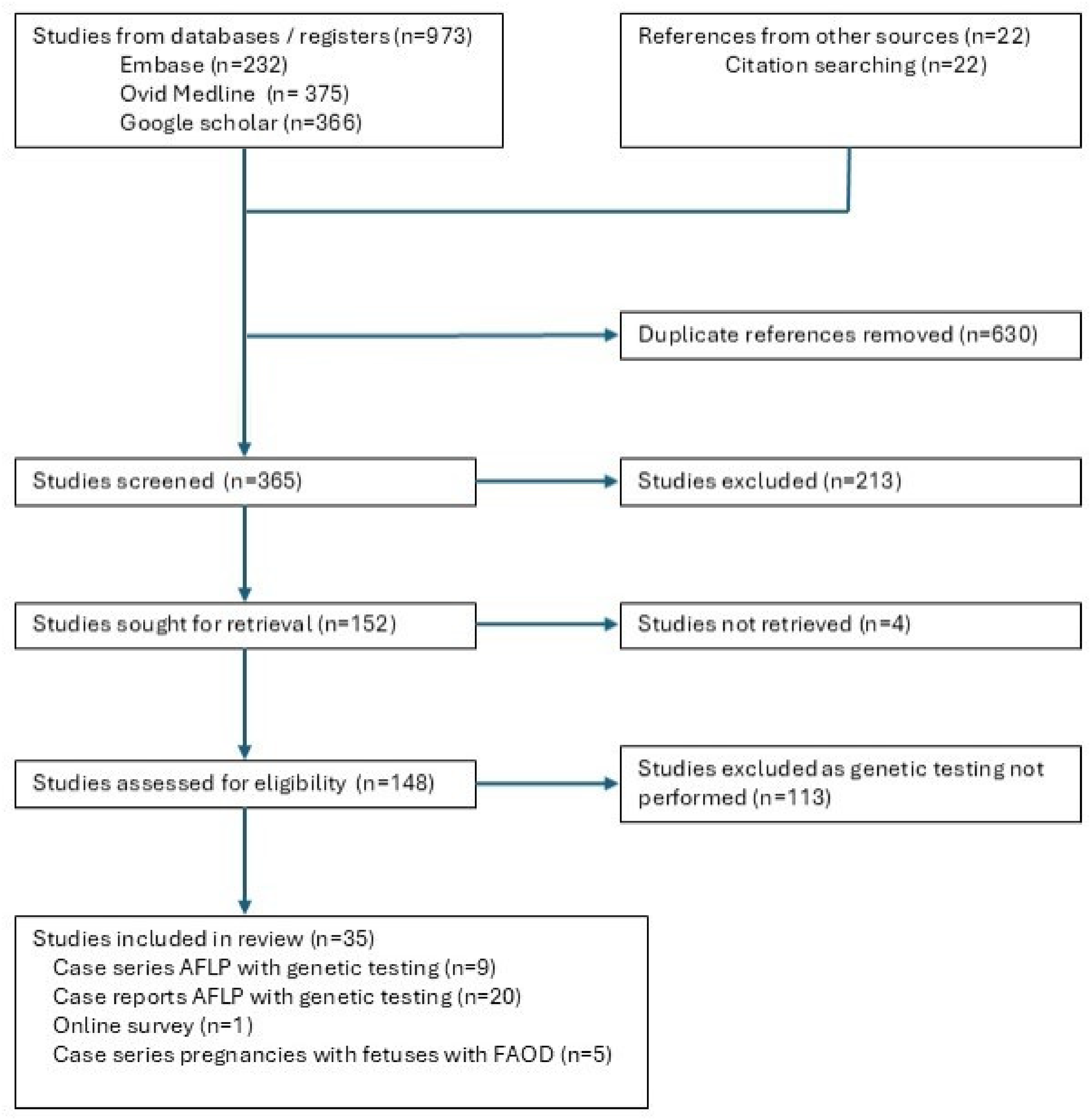
Flowchart of study methods.
Results
The search identified 66 case series comprising 2349 women with a diagnosis of AFLP. Testing for genetic variants was performed in nine case series, totaling 113 pregnancies7–15 (Table 1). Diagnosis of AFLP was based on the Swansea criteria in 69 pregnancies, liver biopsy in 14 pregnancies, previously published criteria in 27 pregnancies prior to 2008, and not stated in 3 pregnancies. Testing of the mother and/or infant demonstrated the common LCHAD c.1528G>C p.(Glu510Gln) pathogenic variant in only five cases (4.4%).
Prevalence of pathogenic variants, abnormal neonatal enzyme analysis or abnormal acylcarnitine profiles in case series of AFLP.
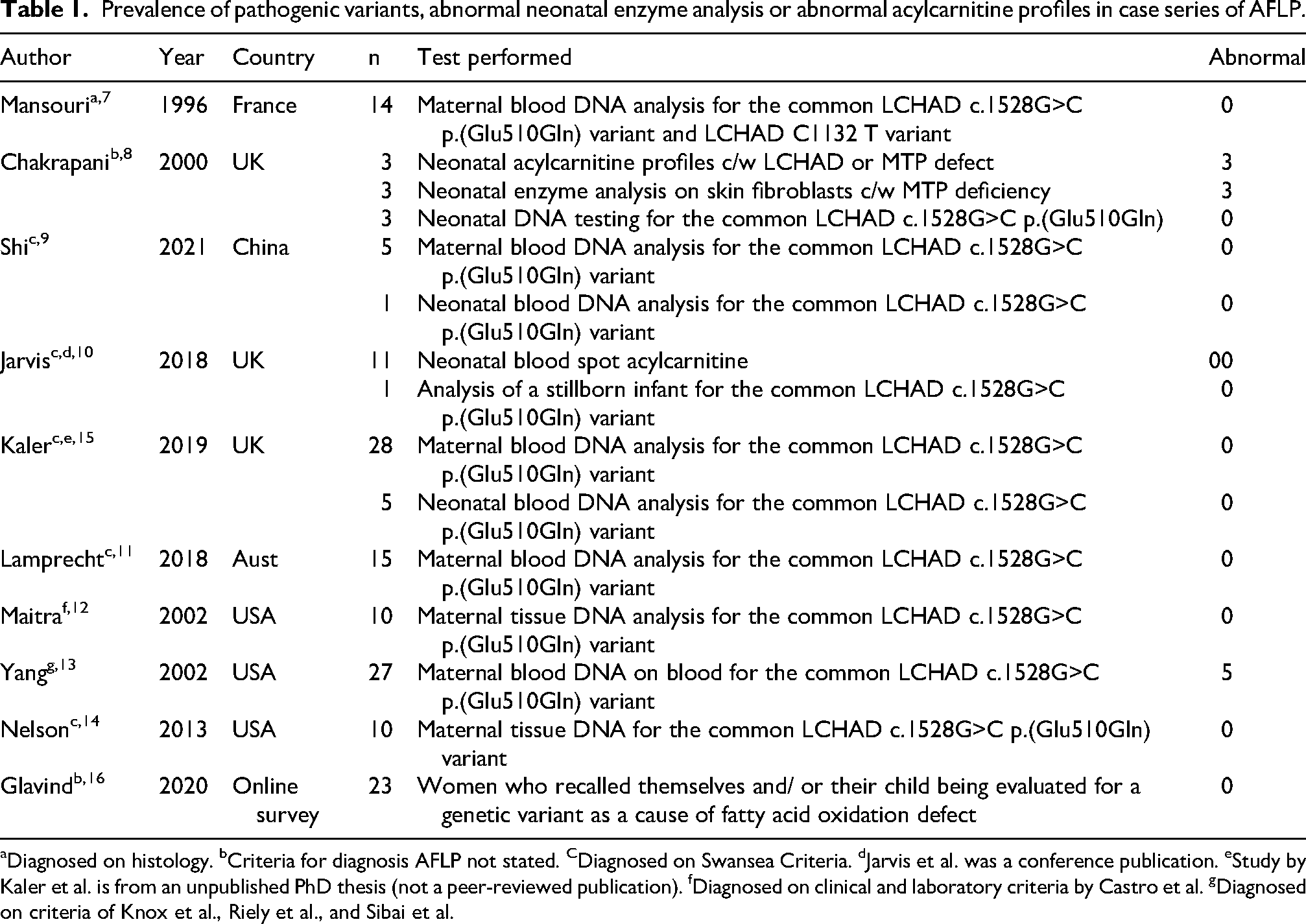
Diagnosed on histology. bCriteria for diagnosis AFLP not stated. CDiagnosed on Swansea Criteria. dJarvis et al. was a conference publication. eStudy by Kaler et al. is from an unpublished PhD thesis (not a peer-reviewed publication). fDiagnosed on clinical and laboratory criteria by Castro et al. gDiagnosed on criteria of Knox et al., Riely et al., and Sibai et al.
An additional online survey found that none of the 23 women with previous AFLP who recalled having themselves and/or their child having genetic testing for a fatty oxidation defect found a pathogenic variant, although the genes tested for were not specified. 16
Seventy-seven case reports were reviewed. Genetic variants were identified in 6 of the 20 case reports (30%) where genetic testing was performed. This consisted of two cases of heterozygosity for the common LCHAD c.1528G>C p.(Glu510Gln) variant, two cases of CPT1, one case of SCAD, and one case of MTP deficiency with a homozygous variant in the HADHB gene.3,4,17–19 No variant was identified in 14 cases.6,18,20–27 LCHAD deficiency was demonstrated in the infants of an additional three AFLP pregnancies; however, genetic analysis was not performed.28,29
Five studies retrospectively examined the prevalence of maternal liver disease complicating pregnancies with a fetus in which a FAOD was subsequently diagnosed. Sims et al. identified three children with LCHAD deficiency who presented with unexplained death or hypoglycemia and abnormal liver enzymes. 30 Two children were homozygous for the common LCHAD c.1528G>C p.(Glu510Gln) variant, the remaining child had the common LCHAD c.1528G>C p.(Glu510Gln) variant on one allele, and a C1132 T mutation creating a premature termination codon (residue 342) on the second allele. The three mothers each had AFLP during pregnancy.
In two papers, the effects of fetal genotype on the pregnancy course in families with mitochondrial trifunctional protein mutations from a cohort were studied.31,32 Twenty-six children diagnosed with LCHAD deficiency, having presented with predominantly hepatic, cardiac, or neuromuscular abnormalities, were identified. Ten children demonstrated a homozygous LCHAD c.1528G>C p.(Glu510Gln) pathogenic variant, while the remaining 16 children were compound heterozygotes, with the common LCHAD c.1528G>C p.(Glu510Gln) pathogenic variant in one allele of the alpha-subunit gene and a different mutation in the other allele. Sixteen of the 26 pregnancies (62%) were complicated by maternal liver disease, 13 with a diagnosis of AFLP and 3 with a diagnosis of hemolysis, elevated liver enzyme, and low platelet syndrome (HELLP). Two other pregnancies were complicated by placental abruption and preeclampsia, respectively. The remaining eight pregnancies were uncomplicated. None of the five pregnancies producing children who subsequently manifested LCHAD deficiency with complete deficiency of the MTP without the common LCHADc.1528G>C p.(Glu510Gln) variant was complicated by maternal liver disease. 31
Browning et al. performed a case-control study examining the prevalence of maternal liver disease during pregnancies where the infant was subsequently diagnosed with a FAOD. 2 Maternal liver disease occurred in three of five pregnancies (60%) with fetuses with LCHAD deficiency. Mutation analysis was performed in two of the three infants, each demonstrating compound heterozygosity with the common LCHAD c.1528G>C p.(Glu510Gln) pathogenic variant in one allele. Maternal liver disease also occurred in 2 of 11 pregnancies (18.1%) with fetuses with SCAD, 2 of 23 pregnancies (8.7%) with fetuses with MCAD, the only pregnancy with a fetus with MTP, and none of 10 pregnancies with fetuses with very long chain acyl-CoA dehydrogenase deficiency.
Tyni et al. described 29 pregnancies with LCHAD-deficient fetuses. Three pregnancies were complicated by HELLP syndrome, one pregnancy was complicated by AFLP, and three pregnancies were complicated by preeclampsia. 33 Homozygosity for the common LCHAD c.1528G>C p.(Glu510Gln) variant was the only mutation demonstrated in affected children, with 10 of the mothers and 6 asymptomatic siblings being heterozygous for this variant.
Discussion
Fetal FAODs are associated with an increased risk of AFLP, of which the most reported is fetal deficiency of LCHAD. 34 LCHAD is part of an enzyme complex, the mitochondrial trifunctional protein (MTP), which is a heterooctamer of 4α- and 4β-subunits, coded by the HADHA and HADHB genes, respectively, on chromosome 2. The α-subunits contain long-chain 3-enoyl-CoA hydratase and LCHAD enzymes, while the β-subunits contain long-chain 3-ketoacyl-CoA thiolase. All 3 enzymes of the MTP are involved in the β-oxidation of long-chain fatty acids in the mitochondria, resulting in the end product of acetyl-CoA, which can then be used for ketogenesis, steroidogenesis, and as a substrate for the tricarboxylic acid (TCA) cycle for ATP production. 35
Defects in the MTP complex are recessively inherited and cause either isolated LCHAD deficiency, with normal or partially reduced thiolase and hydratase activity, or complete MTP deficiency with markedly reduced activity of all three enzymes. The most common pathogenic variant is the LCHAD c.1528G>C p.(Glu510Gln) variant in exon 15 of the α-subunit, which alters amino acid 474 from glutamic acid to glutamine of the HADHA gene, resulting in isolated LCHAD deficiency. 36 More than 30 variants in the HADHA gene have been described to date, and in addition to homozygosity for the common LCHAD c.1528G>C p.(Glu510Gln)variant, compound heterozygosity may result in MTP deficiency. 37 Determination of enzymatic activity is required to characterize isolated LCHAD deficiency or general MTP deficiency. There is significant geographical variation in the frequency of HADHA gene variants, with LCHAD c.1528G>C p.(Glu510Gln) carriers in 1/169 in Poland, 1/680 in the Netherlands, and 0/1200 screened individuals from China. 38
The physiologic metabolic changes during pregnancy result in increased demand for fatty acids. It is hypothesized that the LCHAD-deficient fetus and placenta produce hepatotoxic long-chain 3-hydroxylacyl fatty acid metabolites, which, when combined with the metabolic stress of the third trimester, may cause liver disease in the heterozygous mother.34,35 Defective placental metabolism of long-chain fatty acids likely plays a significant role in the pathogenesis, as the enzymes of fatty acid beta-oxidation are expressed in the placenta, which has the same genetic makeup as the affected fetus. 38 The authors propose that the toxic 3-hydroxy fatty acid intermediates are shunted from the placenta to the maternal circulation to induce AFLP. In women with a history of previous AFLP with negative testing for the common LCHAD c.1528G>C p.(Glu510Gln) variant, Kaler demonstrated absence of ketonuria despite prolonged starvation in the non-pregnant state, suggesting a pregnancy-specific defect in maternal fatty acid oxidation. 15
Chakrapani et al. described three cases of AFLP in which neonatal acylcarnitine profiles and enzyme on skin fibroblasts were consistent with MTP deficiency, though DNA testing for the common LCHAD c.1528G>C p.(Glu510Gln) variant was negative. 8
The presence of a FAOD has important implications because of potential complications for the infant, including hypoketotic hypoglycemia, cardiomyopathy, rhabdomyolysis, rapidly progressing encephalopathy with hepatic dysfunction, and unexpected death. 39 Retinopathy and slowly progressive myopathy and peripheral neuropathy may also occur. 40 The presence of a FAOD may also be important in predicting the risk of recurrent AFLP in subsequent pregnancy. 41 A review of nine case series found a single case of recurrent AFLP in 35 subsequent pregnancies (2.9%), with a further nine cases of recurrent AFLP described in case reports. 41 In addition to DNA testing, measurement of acylcarnitine profiles, total and free carnitine levels, urine organic acids, and enzyme assay in leukocytes or skin fibroblasts may be valuable in infants. Assessment for low levels of serum beta-hydroxybutyrate and urine ketones following prolonged fasting, as well as measurement of acylcarnitine profiles, may be useful in mothers affected by AFLP and the father of the infant. Whole genome testing of the mother, father, and infant of a large series of AFLP pregnancies may be valuable to determine unknown genetic disorders that may predispose to the condition. The low rates of pathogenic variants identified, together with the low rates of recurrence in subsequent pregnancies, warrant greater consideration of non-genetic mechanisms in the pathogenesis of AFLP, including dysfunction of placental metabolic pathways. There is a paucity of information regarding factors predisposing to AFLP. Risk factors for AFLP may include multigravida state, male sex of the fetus, previous episodes of AFLP, and a co-existing diagnosis of other liver disease in pregnancy, including preeclampsia, HELLP syndrome, and intrahepatic cholestasis of pregnancy.23,34,42 In a case series of 17 patients with AFLP, five individuals were noted to have underlying inflammatory bowel disease, and in another, the development of AFLP was preceded by an episode of influenza A hepatitis. 11 The authors proposed that a common feature of these gastrointestinal conditions was the elevation of inflammatory cytokines, which may impair hepatic fatty acid oxidation and induce hepatocyte injury.43–45
Tandem mass spectrometry for acylcarnitine profile is now part of newborn screening in some countries, which can improve early diagnosis of FAOD. Newborn screening specifically increased detection of MCAD deficiency and other FAODs. 46 Treatment of affected LCHAD-deficient infants includes a diet low in long-chain fats supplemented with medium-chain triglycerides (MCT), essential fatty acids, and carnitine. 47 Further research would be required to determine if a similar treatment strategy in pregnant women who are heterozygous for the common LCHAD c.1528G>C p.(Glu510Gln) pathogenic variant would provide any degree of risk reduction for AFLP.
Conclusion
A small minority of women and/or neonates were shown to carry the common LCHAD c.1528G>C p.(Glu510Gln) variant in HADHA in case series of AFLP pregnancies. In the event of negative genetic testing, consideration should be given to additional investigations in the neonate, mother, and father of the baby to identify other fatty acid oxidation disorders. Further research into risk factors and non-genetic mechanisms of AFLP and potential treatments would be valuable.
Footnotes
Ethics approval
Ethical approval was provided by the Mater Health Human Research and Ethics Committee QACR/MML/116423 (V1).
Contributions
JH and AM performed the literature search and wrote the initial draft of the manuscript. JH, AM, LP, and JN reviewed and wrote the final draft of the manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Guarantor
AM.