
Abstract
In the UK over 10 000 people live with cystic fibrosis (CF), with 1-in-25 people being carriers of the disease. Multidisciplinary care is provided by tertiary care CF centres, with or without local secondary service shared care agreements. There are still, however, several reasons why CF sufferers or their families present to their GPs. This article aims to provide a brief overview of CF and its management. It also gives the information needed to guide patients about genetic testing and neonatal screening for the disease.
The RCGP curriculum and cystic fibrosis
The role of the GP in the Clinical topic guide: Genomic medicine is to:
Take and consider family histories in order to identify families with, or at risk of, genetic conditions Identify patients and families who would benefit from being referred to appropriate specialist services Manage the day-to-day care of patients with genetic conditions, even if the patient is under specialist care Coordinate care across services, including transitions from paediatric to adult services Communicate information about genetics and genomics, including discussing results from antenatal and new-born screening programmes Understand how genomic information is used within the context of routine clinical practice Recognise that the identification, assessment, diagnosis and treatment of most acute and chronic respiratory diseases are a primary care issue Consider how respiratory disease affects patients of all ages. It also brings specific challenges in the diagnosis and treatment of various groups including children, some occupational and ethnic groups, those with social and mental health challenges, and those nearing the end of their life Be aware of your role as a GP in promoting smoking cessation and offering treatment Diagnose, investigate and manage digestive symptoms using history, examination, monitoring and referral where appropriate Communicate effectively and consider the social and psychological impact of digestive problems including the potential difficulties for some patients to discuss digestive symptoms Intervene urgently when patients present with emergencies related to digestive health Coordinate care with other organisations and professionals leading to effective and appropriate acute and chronic digestive disease management Offer advice and support to patients, relatives and carers regarding prevention, prescribing, monitoring and self-management The natural history of the untreated condition including whether acute or chronic The prevalence and incidence across all ages and any changes over time Typical and atypical presentations Recognition of normal variations throughout life Risk factors including lifestyle, socio-economic and cultural factors Diagnostic features and differential diagnosis Recognition of ‘alarm’ or ‘red flag’ features Appropriate and relevant investigations Interpretation of test results Management including self-care, initial, emergency and continuing care, chronic disease monitoring, and end-of-life care Patient information and education including self-care Prognosis
The role of the GP in the Clinical topic guide: Respiratory health is to:
The role of the GP in the Clinical topic guide: Gastroenterology is to:
For each problem or disease, consider the following areas within the general context of primary care:
What is cystic fibrosis, and how does it present clinically?
Some of the many manifestations of cystic fibrosis

Red flags for when further testing for cystic fibrosis should be considered.

Cystic fibrosis screening programme
All newborn babies in the UK are offered CF screening as part of the heel-prick test that takes place on day 5. The blood spot obtained is tested for the level of Immunoreactive Trypsin (an enzyme produced in the pancreas). If significantly raised, the sample is genetically tested for the most common CF mutations in Caucasians. The patient is then managed as per Fig. 1. As the testing searches solely for the most common mutations, false negatives can occur, particularly with mutations that are more likely in non-Caucasian groups. Failure to identify a second, but different mutation (not covered by the genetic testing panel), may result in a child being wrongly identified as a carrier rather than a sufferer. Therefore, referrals for further specialist testing should still be made if clinical suspicion is high. Faecal contamination of the sample can result in a false positive test.
Cystic fibrosis immunoreactive trypsin (IRT) screening flowchart.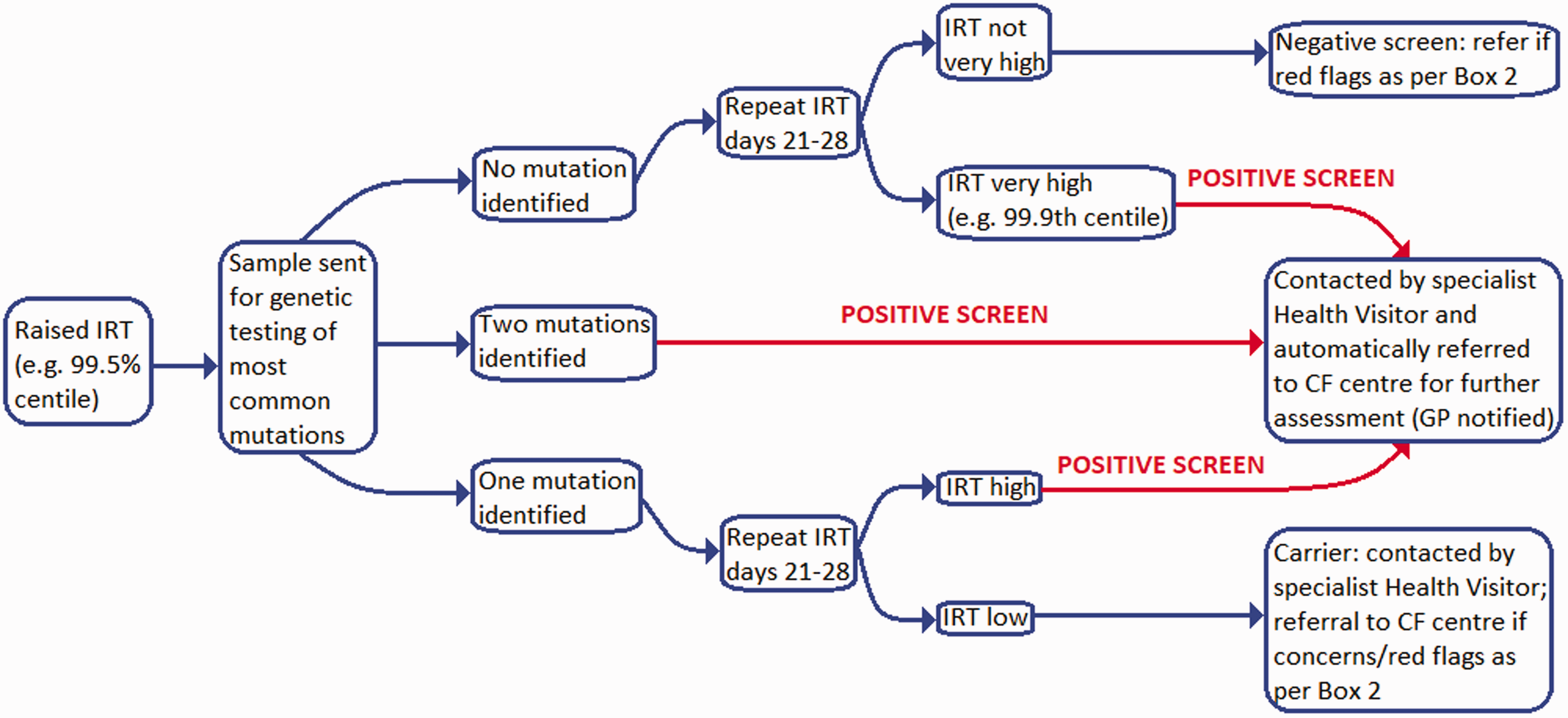
Diagnosis of cystic fibrosis
Diagnosis is confirmed using genetic testing and or a positive sweat test (high chloride concentrations detected). During the sweat test, local sweating is stimulated using pilocarpine-soaked gel pads placed on the patient’s arm or leg, with a small painless current being passed between them. A small collecting duct is then strapped to the area (see Fig. 2) and the collected sweat sent for laboratory analysis. It may need to be repeated if insufficient sweat is collected or the result is equivocal. Occasionally patients may be diagnosed clinically as having CF on the basis of symptoms, even in the presence of an equivocal sweat test and normal genetic testing.
Sweat testing collector in-situ.
The genetics: Who should be screened?
With CF being a recessive disorder, both parents need to be carriers for a child to be born with the disease (see Fig. 3). With two carrier parents there is a 25% chance that each of their offspring will have CF.
Diagram showing genetic inheritance of cystic fibrosis.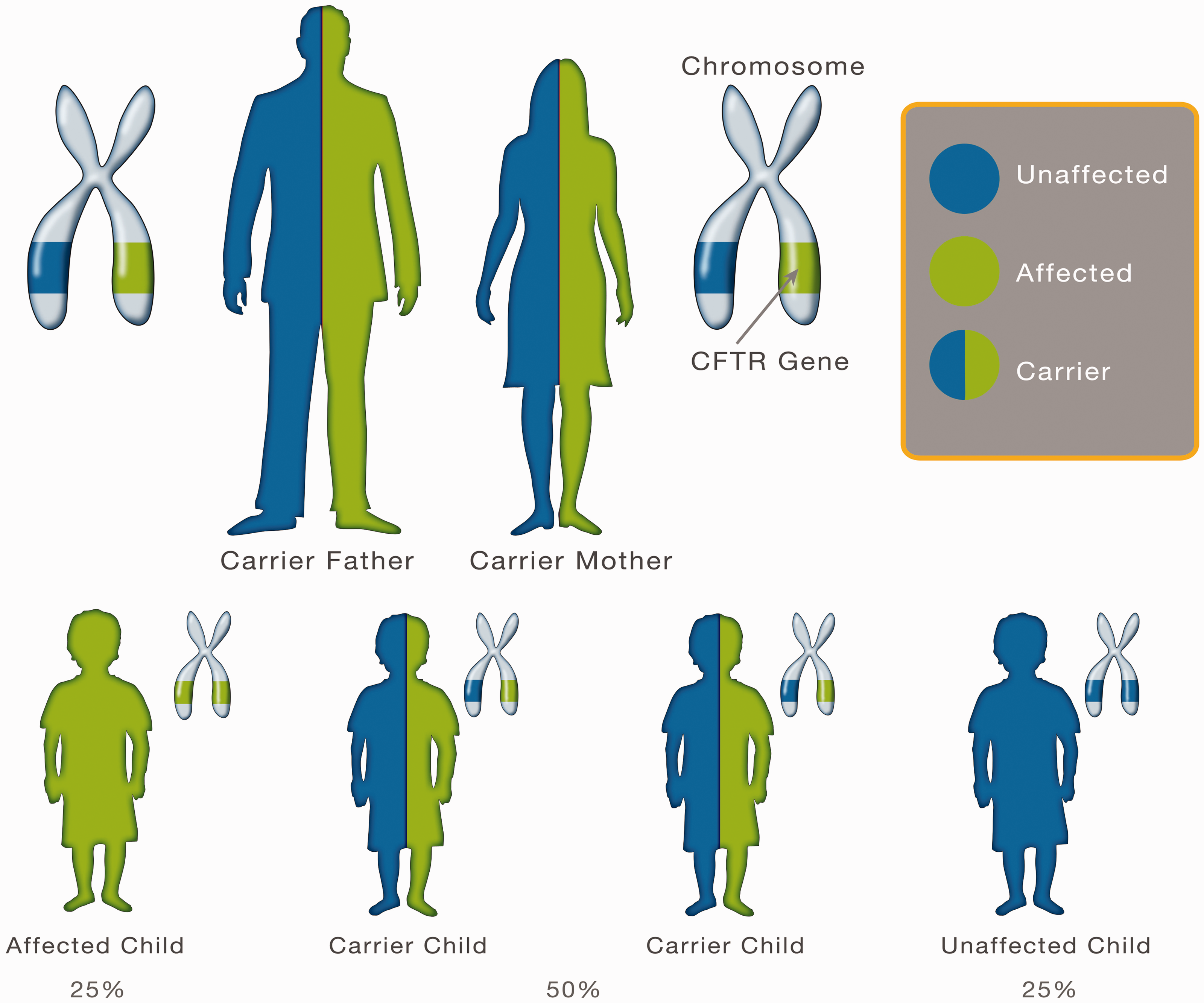
If an individual with CF (the index case) is identified, then cascade testing of other family members is offered. A sibling of an affected individual has a two-in-three chance of being a carrier (if sharing the same parents) and assuming that they themselves are not affected. Other close relatives may ask to be tested. Further advice on testing and who to test can be obtained from local genetic testing centres.
After explaining inheritance, some individuals may not wish to know their CF genetic status and opt out of further testing. For other individuals, confirmation of their genetic status may guide future reproductive planning. Patients, when appropriate, should be referred to genetic services for further genetic counselling and testing. If the gene mutation of the index case has been identified, then the family member can be tested. As partners are only screened for the common mutations, there is a chance that a false negative result will occur if they carry a rarer mutation.
Options for future pregnancies
If both partners are found to be carriers, the implications for future pregnancies need consideration. Some may opt not to start a family, whereas others may choose to use donor gametes or adopt. Pre-implantation genetic diagnosis as part of In vitro fertilisation (IVF) is a further option, although funding for this varies. Pre-implantation genetic diagnosis involves genetic profiling of embryos in the IVF process, with unaffected embryos then selected for implantation. Patients who are either pregnant or who wish to conceive naturally have the option of genetic testing via chorionic villous sampling or amniocentesis. Although both procedures carry a small, but significant, risk of miscarriage, they offer parents an opportunity to prepare for, or if preferred terminate the pregnancy. Non-invasive prenatal testing, by testing maternal serum for fetal DNA is offered by the NHS in some areas.
Cystic fibrosis management
Respiratory disease sequalae
CF patients have thickened respiratory secretions, reduced mucocilliary clearance, and an exaggerated inflammatory response to infection. They are susceptible to recurrent infections with organisms such as Staphylococcus aureus, Pseudomonas aeruginosa and Burkholderia cepacia; cycles of infection and inflammation result in bronchiectasis and progressive lung damage, with respiratory failure remaining the most common cause of death in patients with CF (Sorde et al., 2011). Treatments aim to improve mucocilliary clearance, clear infection and slow the progression of lung damage. Patients require regular specialist review, with chest radiographs and spirometry as part of a host of assessments. Progression of the disease may result in the patient needing non-invasive ventilation, oxygen and ultimately lung transplantion.
Despite aggressive management with antibiotics, patients unfortunately, usually become colonised with bacteria. Paeruginosa colonisation is associated with worsening lung function and increased mortality (Emerson et al., 2002). It is managed with long-term nebulised or inhaled antibiotics, for example colistimethate (Colistin) or, if ineffective, tobramycin (local funding criteria apply for some preparations), which can improve lung function and reduce hospital admissions (Ramsey et al., 1999). Azithromycin used at specific immunomodulating dosages can also improve lung function and reduce hospital admissions in patients colonised with P.aeruginosa (Saiman et al., 2003; Sorde et al., 2011). B.cepacia infection, another poor prognostic indicator (Folescu et al., 2015), is managed according to microbiological advice.
Chest physiotherapy in varying forms is required from once to multiple times daily; increasing frequency is required in co-morbid infective exacerbations. Different techniques are taught, depending on both age and individual factors, and techniques can be self or carer administered.
Oscillating vests/devices may be used or purchased by patients, although evidence suggests they may be less effective than the therapeutic approaches above for compliant patients (National Institute for Health and Clinical Excellence (NICE), 2017). Compliance with physiotherapy is fundamental, and may be difficult to ensure at high-risk times such as during adolescence or when there is a co-morbid deterioration in psychological wellbeing. Exercise is thought to be beneficial to lung function, and should be generally encouraged (NICE, 2017).
The mucolytic Dornase alfa (a genetically engineered version of an enzyme that breaks down DNA in respiratory secretions, thus reducing viscosity) is used in appropriate patients with evidence of lung disease. In systematic reviews, it has been shown to reduce exacerbation frequency and the rates of decline in lung function (Yang and Montgomery, 2018). Hypertonic saline nebulisers may be used alone or in addition to the above, and Mannitol dry powder may be used in patients when the above treatments are ineffective or unsuitable (NICE, 2017).
Sputum cultures (or cough/nasal pharyngeal aspirates if more practical) should be checked regularly. Positive cultures should prompt aggressive management with antibiotics, irrespective of whether clinical symptoms are present or not. As the pharmacokinetics of antibiotics in CF patients may differ from those of the general population, antibiotic doses and regimens may differ from those normally prescribed. Infections identified in primary care should, therefore, be discussed with the patient’s CF specialist, or administered as per an agreed shared care protocol. Parents of CF children, and when older, the patients themselves, become expert at managing CF, and often recognise when they should liaise with their specialist directly. GPs should otherwise have a very low threshold for discussion with the specialist. Even CF children presenting with what appears to be a ‘simple viral upper respiratory tract infection’ may require antibiotics, additional treatment or monitoring from their specialists, and further advice should be sought.
From diagnosis, babies are usually started on flucloxacillin for S.aureus prophylaxis. Evidence of benefit for other prophylactic regimens is variable, and tends to be individually determined based on disease characteristics, severity and the organisms identified. If organisms are detected, they are treated aggressively in the first instance with intravenous (IV), inhaled or nebulised antibiotics. If longer-term IV antibiotics are required patients may benefit from vascular access devices.
Cross-contamination of colonised bacteria between CF sufferers must be avoided. On a social level, CF sufferers are advised against meeting other sufferers to avoid cross-infection and clinics and hospital admissions locales are adapted to avoid colonised patients mixing with those who are non-colonised (NICE, 2017). Most CF patients should receive the Flu vaccine, and this should be offered to carers. Patients should also receive pneumococcal vaccination. GPs should seek advice if patients with CF develop varicella-zoster as this is associated with an increased risk of respiratory complications.
Pancreatic disease sequalae
Pancreatic insufficiency (diagnosed by measuring faecal elastase) is found in 85% of patients (Singh and Schwarzenberg, 2017) with CF and is associated with a worse morbidity and mortality. It results in malabsorption, particularly of fats, and as a consequence the fat-soluble vitamins A, D, E, and K. Clinically this may manifest as a failure to thrive or steatorrhoea and can result in rectal prolapse. Dietitian support is paramount. Patients are started on A, D, E, and K vitamin supplements and high-calorie diets, in addition to pancreatic enzyme replacement (pancreatin, e.g. Creon), taken with each meal or snack. Compliance with diet and enzymatic replacement may become an issue in adolescents for whom body image concerns and peer pressure may become a hindrance. Some patients require supplementary feeding via gastrostomy if unable to maintain and balance their higher energy requirements.
Involvement of the endocrine part of the pancreas can result in CF-related diabetes. This is a discrete entity, although it shares features of both type-1 and type-2 diabetes (Moran et al., 2010). It is treated with insulin replacement, but a high calorie diet should be maintained. Patients older than 10 years are usually screened for this yearly with oral glucose tolerance testing according to local policies. Haemoglobin A1c (HbA1c) is often low in patients with CF and is not routinely used for screening.
Liver disease sequalae
Cystic fibrosis-associated liver disease (CFLD) affects up to 30% of the CF fibrosis population (Kobelska-Dubeil et al., 2014). Secretive obstruction within the hepatobiliary system can result in gallstones, cirrhosis and ultimately portal hypertension and liver failure. Ursodeoxycholic acid is routinely used to treat and prevent the progression of CFLD. It is safe and well-tolerated, although evidence of benefit is very limited (Cheng et al., 2017). In view of a lack of alternative treatment for CFLD, use of ursodeoxycholic acid still remains in guidance (NICE, 2017). Ultimately, liver transplantion may be required with disease progression.
Bowel disease sequalae
Meconium ileus (bowel obstruction due to abnormally ‘sticky’ meconium) occurs in 20% of patients with CF (Sathe and Houwen, 2017). As a result of the altered electrolyte, water and molecular content of their faeces, CF patients also suffer with chronic constipation requiring aggressive treatment with laxatives. They are also at increased risk of intussusception and the common complication of distal intestinal obstruction syndrome (DIOS). DIOS occurs when thick sticky mucous in combination with viscoid faeces obstructs the bowel (Lavie et al., 2015). It should be suspected if there is acute onset of periumbilical pain associated with bowel obstructive symptoms and a palpable mass in the right lower quadrant. Patients presenting with these symptoms require urgent hospital admission.
Other cystic fibrosis manifestations
Patients with CF are at increased risk of osteopenia and osteoporosis for a variety of reasons including malabsorption, vitamin deficiency, reduced levels of exercise, and from medication side effects (Kerem et al., 2005). Dual-energy X-ray absorptiometry monitoring may be required. CF patients are at increased risk of arthritis and hypertrophic pulmonary osteoarthropathy (digital clubbing with inflammation of the periosteum of long bones). Chronic rhinosinusitis and polyps are also common and treated as in non-CF patients.
Reproductive impact
Most men with CF have azoospermia secondary to the absence of a vas deferens. Intracytoplasmic sperm injection IVF may be an option to enable formation of a zygote. This involves sperm being aspirated directly from the testes. Women with CF quite often have normal fertility, but will require close monitoring of lung function and other parameters during pregnancy (Kerem et al., 2005). They would have the same risks of genetic transmission as male suffers.
Psychosocial Impact
Useful contacts for patient and family support.

Palliative care and organ transplantation
Palliative care teams have much to offer patients and their families, and their input can be invaluable, particularly in the later stages of disease. Although organ transplantation may be an option for some patients, many do not fit the criteria, with some too unwell for transplantation surgery. For others a suitable organ may not be available in time.
Prognosis and the future
With the input from specialist CF centres, life expectancy is continuing to improve. According to the UK CF registry, half of CF patients born in 2017 are expected to live until at least age 47 years. With newborn screening, earlier detection and thus earlier treatment this figure may improve further. Research is ongoing and may identify further gene therapies. Ivacaftor, a licenced therapy for patients with a specific (unfortunately, uncommon) form of CFTR gene mutation, is now available. Other gene-targeted therapies are on the horizon.
KEY POINTS
Patients with CF are managed predominantly by specialist tertiary care CF centres and require multidisciplinary input GPs should seek CF team specialist advice or follow shared care agreements regarding antibiotic prescribing for positive sputum samples A normal or carrier-status on neonatal heel prick screening does not completely exclude CF Red flags warrant consideration of referral for CF exclusion and these include: Persistent cough, failure to thrive, steatorrhoea, and nasal polyps or rectal prolapse in children Compliance with treatments is paramount and may be reduced during high risk times such as adolescence when extra support and encouragement may be required Having a child with complex medical needs can impact significantly on family dynamics and the wellbeing and health of other family members
Footnotes
Acknowledgement
We thank Dr Elizabeth Campy ST8 paediatrics, University Hospital North Durham, for proofreading this article.