
Abstract
Genetic haemochromatosis (GH) is the UK’s most common genetic condition inherited in an autosomal recessive manner. It is an iron overload disorder, in which increased intestinal absorption of iron leads to deposition of iron in organs and tissues, most commonly the liver, heart, pancreas, and joints. GH often presents in an insidious manner, and as a result is significantly underdiagnosed. It is a multi-system disease, in which patient care involves various specialities, including primary care. This article will outline the inheritance, diagnosis, treatment and implications of GH and aims to raise awareness of the condition in general practice.
The RGCP curriculum and haemochromatosis
The role of a GP in the haematology and genomic medicine clinical topics is to:
Identify symptoms that are within the range of normal or self-limiting illness and differentiate them from underlying pathology e.g. anaemia Know the epidemiology of common disorders and understand how to recognise them Make an effective assessment, including conducting more detailed tests and referring appropriately Take and consider family histories in order to identify families with, or at risk of, genetic conditions (including autosomal and X-linked disorders) and familial clusters of common conditions such as cancer, cardiovascular disease and diabetes Identify patients and families who would benefit from being referred to appropriate specialist services Manage the day-to-day care of patients with genetic conditions, even if the patient is under specialist care Coordinate care across services, including transitions from paediatric to adult services Communicate information about genetics and genomics, including discussing results from antenatal and new-born screening programmes Understand how genomic information is used within the context of routine clinical practice The natural history of the untreated condition including whether acute or chronic The prevalence and incidence across all ages and any changes over time Typical and atypical presentations Diagnostic features and differential diagnosis Appropriate and relevant investigations Interpretation of test results Management including self-care, initial, emergency and continuing care, chronic disease monitoring Patient information and education including self-care Prognosis
The RCGP curriculum clinical topic guide details emerging issues and the knowledge and skills guide that may be relevant when encountering haematological conditions and genomic medicine. This article will cover the following areas:
Introduction
Genetic haemochromatosis (GH) is the most common genetic condition in people of European descent (Pilling et al., 2019). The p.C282Y variant is present in 10–15% of populations of northern European descent, with approximately 1 in 150 people being homozygotes and is the highest genotype prevalence reported in Ireland and Britain (Bomford, 2002). Professor David Melzer and the Exeter Haemochromatosis Research Group proposed that there may be 380 000 people affected in the UK (Pilling et al., 2019). However, the condition is significantly underdiagnosed, with the NHS being aware of just 14 000 people with GH (Smith et al., 2016). As such, a GP list of 2000 patients will have approximately four patients with GH. Despite this point, many GPs have never seen a case, although they are often best placed to make a diagnosis (Sood et al., 2013).
Diagnosed early, GH is manageable and not life-limiting, and treatment is simple and cost-effective. Recent studies based on UK Biobank data suggest that each year in the UK, GH is responsible for 564 new patients with liver disease and 125 new liver cancer patients (Pilling et al., 2019). According to Cancer Research UK, 5-year survival rates for liver cancer can be as low as 2%, depending upon age at diagnosis and gender. Therefore, delayed diagnosis significantly increases the disease burden for patients and the costs to the NHS. We hope that this article will raise awareness of the diagnosis and management of GH to enable more sufferers to be diagnosed earlier and managed appropriately.
Diagnosis and investigation
Symptoms related to genetic haemochromatosis.

If a clinician feels that any one of the listed symptoms or signs is present without another clear cause, we should consider the possibility of GH and investigate appropriately. In general practice, we typically see patients with concerns about one or two issues. In some cases, these conditions may seem unrelated and it may only be after a thorough review of a patient’s notes that a pattern becomes evident.
History
Some patients may present because they have a family history of GH and are wondering if they have inherited this illness. In this case first line investigations should be initiated, as the potential benefit of an early diagnosis would outweigh any risk of taking a blood sample, even if diagnosis based on the history and examination seems unlikely.
The majority of patients will present with non-specific symptoms that may be challenging to interpret. Some key areas to ask about are:
General health and energy levels Changes in mood, memory or increased irritability Changes in skin colour or hair distribution Changes in libido or sexual impotence in men Menstrual history including any unusual patterns of menstruation
Examination
As the symptoms and presentation can be so varied, at the initial consultation, there may be a very limited examination focussing on the presenting complaint (e.g. joint pain) or no examination at all. Examination may occur after or alongside investigation findings that may direct further examination. The main physical manifestations to detect on examination are listed in Box 2 (Mohamed and Phillips, 2016) and depicted in Fig. 1.
Organ systems that can be affected by genetic haemochromatosis. Physical findings in genetic haemochromatosis.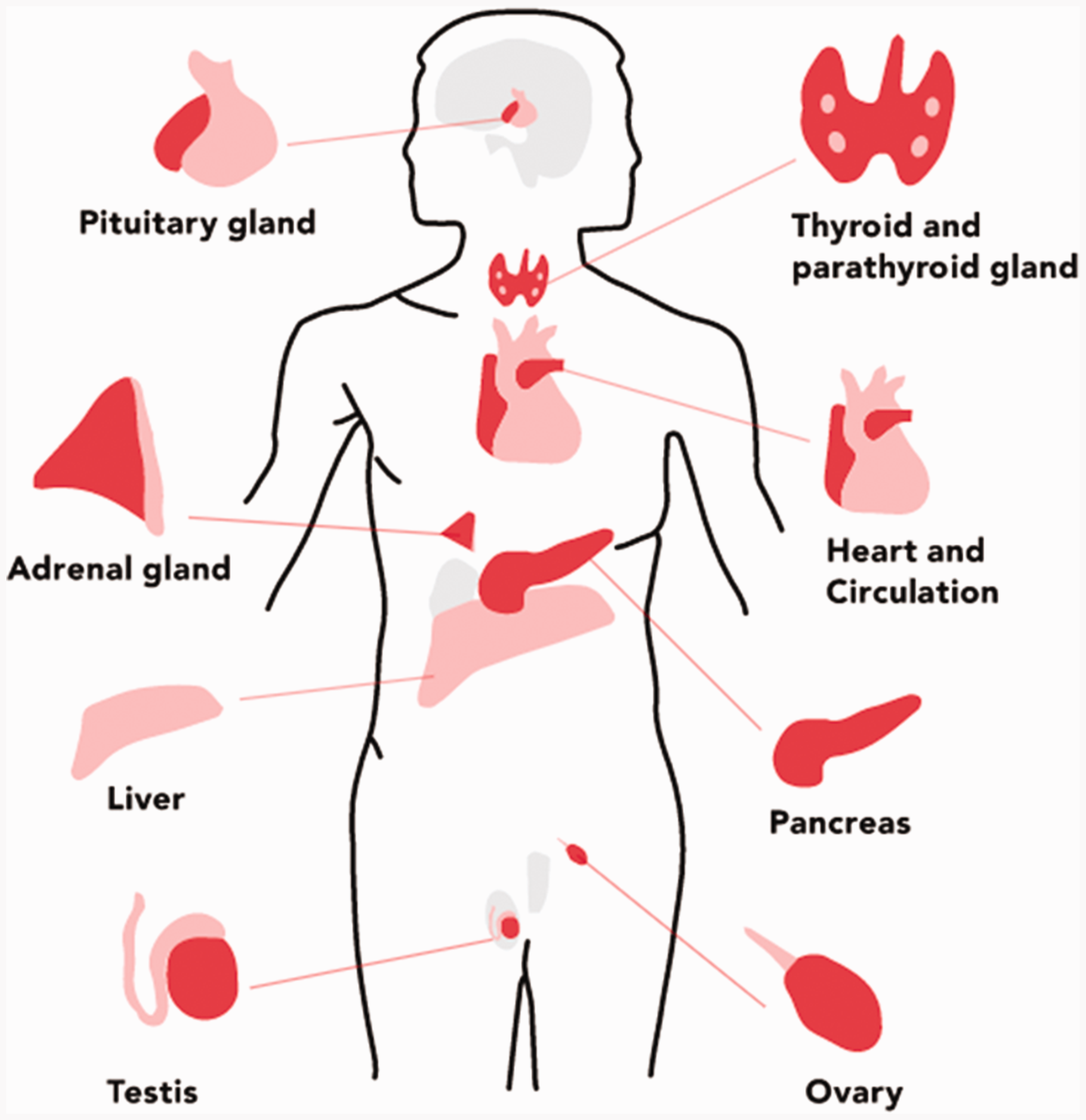

Investigations
British Society of Haematology diagnosis of genetic haemochromatosis.

If these results are present we should refer to haematology or gastroenterology, depending on local guidelines, for further management. If the patient does not quite meet the thresholds, or if there are concerns about any results, these should be discussed with a specialist before deciding on the next step.
In reality, in primary care we are often prompted to consider GH as a cause of symptoms when we get a raised SF result. Patients found to have raised SF should be asked about alcohol intake and other risk factors for liver disease. Transfusion history, family history of iron overload, history of type 2 diabetes mellitus, obesity and hypertension, as well as for symptoms or signs of underlying inflammatory or malignant disorders (Cullis et al., 2018). These can all be causes of a raised SF and if present in a patient’s history, then we should react to this information appropriately and investigate further as needed. Only a very small number of people with raised SF actually have GH, as there are many other reasons SF can be raised. Figure 2 is from an article in the British Journal of Haematology and provides a structured method in which to investigate raised SF (Cullis et al., 2018).
Management of raised serum ferritin.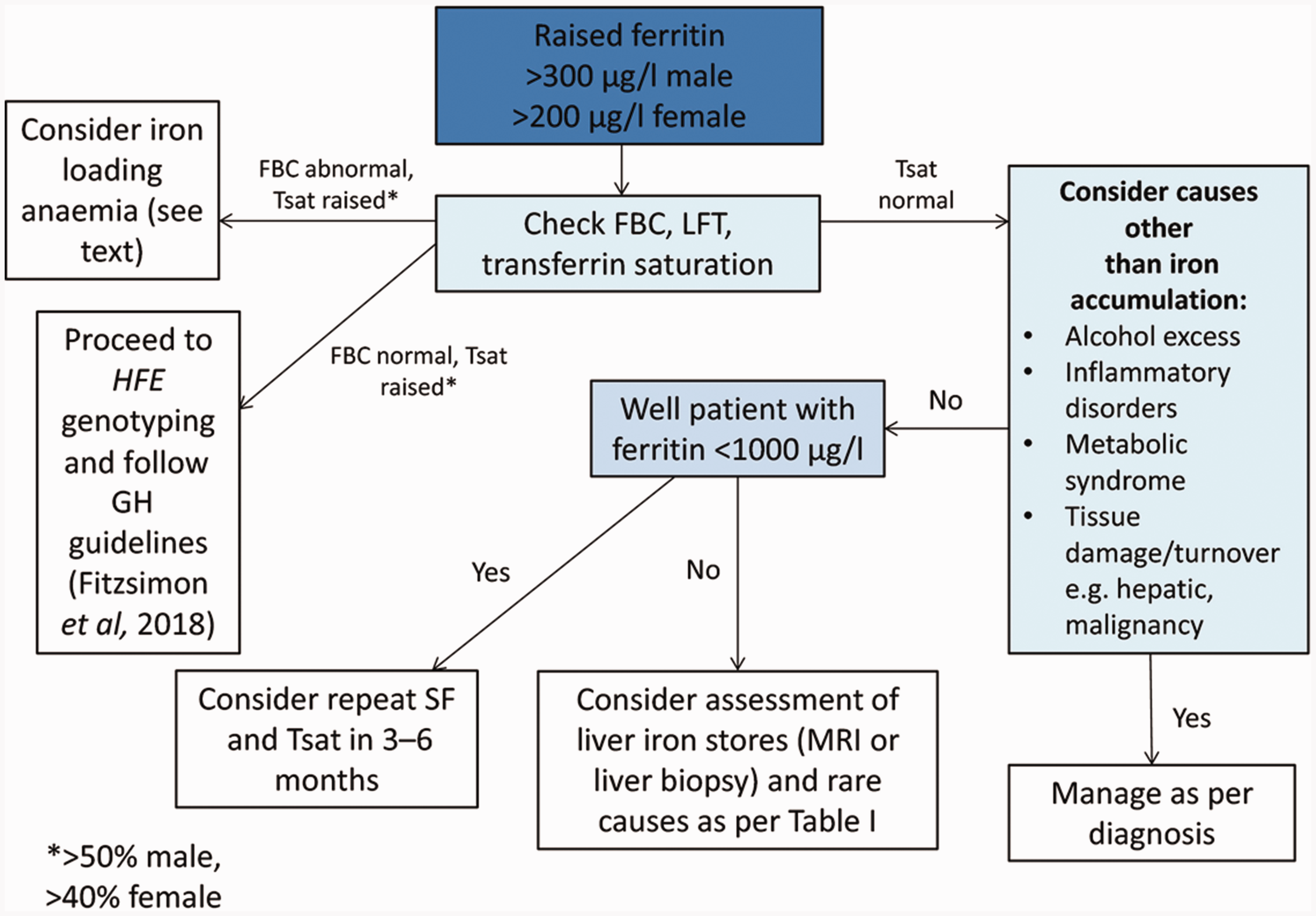
Ferritin and transferrin explained.

Genetics
The different types of haemochromatosis.

Over 90% of patients with GH carry a mutation of the human-leucocyte antigen class-I-like gene, (HFE) at amino acid 282 (C282Y). There is a second variant at amino acid 63 (H63D). The second mutation is more common in the general population, but is not usually associated with iron accumulation (Fitzsimmons et al., 2017). The HFE gene produces a transmembrane glycoprotein. Through complicated interactions with other receptors and proteins this protein reduces hepcidin expression in hepatocytes in GH Type 1.
Hepcidin stops the release of iron from enterocytes and macrophages by causing the breakdown of the iron exporter, ferroportin. Hepcidin levels are reduced in Type 1 GH, and therefore, more iron is released from enterocytes and macrophages. As this cannot all be stored in the usual places (red blood cells, myoglobin, etc.) storage occurs in other places including liver, pancreas, joints and heart. Iron intake remains the same whereas total body iron increases from 4 g in a normal individual to 20 g in a person with GH, and this causes the physical manifestations of GH as there is no regulatory pathway for the excretion of excess iron.
It is important to note that even though a patient may be a homozygote for one of the genetic changes described above in Type 1 GH they will not necessarily go on to have GH clinically. This is because the clinical penetrance is variable depending on which exact copies of the HFE gene a patient has inherited and the mixture of C282Y and H63D mutations that are present in these genes.
Management
As per the RCGP curriculum guidance, management of GH can be divided into self-care, initial management, continuing care and chronic disease management. As GH is a multi-system disease the management is likely to involve several specialities.
Self-care
Self-care focuses on diet and lifestyle modification. It is important to advise patients that changes to diet cannot prevent iron overload, but making certain changes can affect the rate at which patients accumulate iron. The following advice is recommended by the British Liver Trust in the information section of their website (British Liver Trust, 2018):
You should avoid consumption of the following:
Vitamins or multivitamin supplements that contain iron Vitamin C in pill form as this increases absorption of non-haem iron. Vitamin C from fruit and vegetables does not need to be avoided Breakfast cereals that are 'fortified' with iron Shellfish such as oysters, mussels and clams as these contain a bacteria that may be fatal to people with iron overload Because of the increased absorption from animal foods you may wish to cut down on eating red meat. Offal (organs such as heart, liver, kidneys etc.) in particular is very iron-rich There are certain substances that should be included in your diet: Calcium, as found in dairy foods, limits the absorption of haem iron (it is therefore helpful to consume dairy foods when you are eating meat) Tannin, as found in tea, limits the absorption of iron
Uncooked seafood should be avoided, due to the increased risk of infection with Vibrio vulnificus in these patients (Adams and Barton, 2010). V.vulnificus is a gram negative bacillus that appears to grow rapidly in the blood of people with GH (Bullen et al., 1991). It has been associated with sepsis and cellulitis.
As GH can increase a patient’s risk of developing liver disease and diabetes, affected individuals should be advised to take steps to modify other risk factors associated with these diseases, namely maintaining a healthy weight and moderating alcohol intake. Consumption of more than 60 g of alcohol per day has been associated with substantially higher levels of serum iron and ferritin (Powell et al., 2016). Fletcher and colleagues described a nine-fold increase in cirrhosis in C282Y homozygote patients who drank more than 60 g of alcohol per day compared with those who drank less than 60 grams of alcohol per day (Fletcher et al., 2002).
Initial management
As part of their initial management, all patients should be referred to a gastroenterologist or haematologist depending on local guidelines. Management will then depend on whether the initial SF level was less than 1000 microgram/L or greater than 1000 microgram/L. GH patients who present with SF greater than 1000 microgram/L and any with raised transaminases should be referred to a hepatologist for fibrosis assessment and exclusion of cirrhosis and that liver biopsy is no longer required for diagnosis of HFE GH, but may be required to assess the severity of fibrosis in GH patients with SF greater than 1000 microgram/L and or elevated transaminases. Transient elastography could be used to select which patients from this group require liver biopsy (Fitzsimons et al., 2017).
The patient should be advised to inform first degree relatives so that they too can be screened for GH. FBC, LFTs, SF, TS and HFE should be offered to family members after the diagnosis of HFE GH (Fitzsimons et al., 2017). Family screening should include parents (if available), siblings, partner and children (over the age of consent). Extended family screening is not recommended if an individual is identified as a C282Y/H63D compound heterozygote.
The sibling of an affected patient will have a 1 in 4 chance of also having inherited GH HFE, thus genotyping of the spouse is invaluable and can guide the need for testing of children in later life (European Association for the Study of Liver, 2010). Below the age of consent, it is rare for significant iron overload to have occurred, and so it is not necessary to subject young children to blood tests. They should also be able to give informed consent prior to being tested for a disease, which as mentioned before, may never cause clinical features.
Continuing care
The continuing care element of GH focuses on removing the body’s excess iron to prevent end organ damage. The commonest form of this involves regular venesection. This treatment is split into an initial ‘de-ironing’ stage and then a maintenance phase. By removing blood, the body uses some of its stored iron to make new red blood cells, ultimately depleting the excess iron stores.
Venesection prevents further tissue damage, reduces fatigue, improves skin hyperpigmentation, and improves early hepatic fibrosis and left ventricular dysfunction (Bacon et al., 2011). During a venesection up to 500 mL of blood will be removed in one procedure, this equates to about a quarter of a gram of iron (Adams and Barton, 2010). The frequency of venesection during the de-ironing phase can be tailored to the individual patient and depends on factors such as the degree of iron overload at presentation, comorbidites, age and the patient’s ability to tolerate the procedure without side effects. Initially this may be as frequently as weekly.
At diagnosis, all fit GH patients with biochemical iron loading should undergo weekly venesection until SF 20–30 microgram/L and TS less than 50%. During this phase of venesection FBC should be monitored weekly and SF TS monitored monthly. Homozygotes with normal iron indices and compound heterozygotes with minimal elevation of iron indices may be suitable for blood donation and annual monitoring of SF and TS (Fitzsimons et al., 2017).
Once the iron stores have been depleted and serum ferritin is less than 50 micrograms/L the patient can enter the maintenance phase of treatment. During maintenance, patients should undergo venesection as required, preferably at a blood donation centre, to maintain normal FBC with SF less than 50 microgram/L and TS less than 50% (Fitzsimons et al., 2017).
The Joint United Kingdom (UK) Blood Transfusion and Tissue Transplantation Services (XXXX) Professional Advisory Committee (JPAC) guidelines state that:
The selection criteria/methods for all donors with GH preserve the principles of altruism Blood donated for therapeutic use by any donor known to have GH meets all other criteria (except donation frequency) in the JPAC Donor Selection Guidelines. If it is clinically necessary for individuals to donate more frequently than the minimum donation interval, specific permission must be obtained from the designated clinical support officer The donor is under the continuing care of a physician who is able to offer alternative venesection facilities whenever, for any reason, the donor does not meet all other criteria in the JPAC Donor Selection Guidelines.
In patients who cannot tolerate venesection, it may be necessary to use iron chelation therapy. There is limited experience of using the oral iron chelator deferasirox (Exjade) in patients with C282Y homozygous GH and modest iron loading (median SF 645 microgram/L). SF values were modestly reduced, but significant patient withdrawal from gastrointestinal, renal and hepatic toxicities was observed (Phatak et al., 2010).
There is some evidence to suggest that the use of proton pump inhibitors (PPIs) can reduce absorption of iron. In patients with GH PPIs can inhibit the absorption of non-haem iron from a test meal and the habitual diet (Hutchison et al., 2007).
Chronic disease management
Chronic disease management is centred on the prevention of complications, particularly in those patients who have been found to have cirrhosis. These patients have a 100-fold risk of developing primary liver cancer.
Patients with HFE GH complicated by cirrhosis should also be treated to a SF less than 50 microgram/L and TS less than 50%, but they are not suitable for blood donation. We should monitor alpha feto-protein and hepatic ultrasonography every 6 months (Fitzsimons et al., 2017).
As the risk of hepatocellular carcinoma (HCC) is not mitigated in patients with cirrhosis even after adequate phlebotomy, screening for HCC must continue lifelong (Mohamed and Phillips, 2016). Other investigations that may be necessary are electrocardiograph, echocardiography, luteinising hormone, serum testosterone, follicular-stimulating hormone and TFTs if endocrine organ damage is suspected (Bacon et al., 2011). All patients with GH are at an increased risk of osteoporosis, and should have a dual energy X-ray absorptiometry scan (European Association for the Study of Liver, 2010).
Prognosis
People with untreated haemochromatosis who express the clinical phenotype have a higher risk of mortality than the general population. A longitudinal cohort study of 179 patients with haemochromatosis showed reduced survival compared with a matched normal population (P<0.0001) (Milman et al., 2001).
Once cirrhosis and diabetes mellitus have developed, patients have a shortened life expectancy, and if cirrhosis is present, a high risk of liver cancer even when iron depletion has been achieved. If phlebotomy is started before the onset of cirrhosis and diabetes, the prognosis is normal (Fitzsimons et al., 2017).
Information for patients
Information sources for patients.

Quality improvement ideas
The RCGP has teamed up with Haemochromatosis UK and produced an online learning module. This can be accessed via the RCGP Learning website. Complete this module to add to your ePortfolio as a log entry in addition to reading this article.
Audit male patients in your practice who have a raised SF greater than 300 micrograms/L and female patients whose SF is greater than 200 micrograms/L. How many have had the appropriate investigations to exclude GH as a cause of their raised SF?
KEY POINTS
GH is the most common genetic condition in people of European descent It is significantly underdiagnosed with only 14 000 out of a predicted 380 000 cases known to the NHS in the UK Early diagnosis can enable sufferers to have a normal prognosis, but sufferers report high levels of symptoms including fatigue, joint pain and cognitive/psychological issues Presenting symptoms and signs can be vague, making early diagnosis difficult The mainstay of treatment is venesection, moderating alcohol intake is also important Left untreated, sufferers have a much greater risk of developing cirrhosis and HCC