
Abstract

What is bullous pemphigoid?
Bullous pemphigoid (BP) is the most common autoimmune blistering skin disease and typically affects the elderly. There are 3000 new UK cases each year with an incidence of 4.3 per 100 000 person years (Langan et al., 2008). BP can be associated with considerable morbidity and increased mortality owing to secondary infection, sepsis and adverse effects of treatment (Miyamoto et al., 2019). Therefore, it is important for general practitioners to recognise and manage it acutely and refer urgently to a dermatologist.
What is the underlying cause of bullous pemphigoid?
BP is an immunobullous condition where blister formation occurs as a result of pathogenic autoantibodies, usually of IgG and IgE type, which attack proteins (namely BP180 and BP230) within the hemi-desmosomes, which are molecular bridges between the epidermis and dermis layers of the skin. This results in sub-epidermal blister formation (Oakley, 2016).
What are the risk factors?
BP typically presents in the elderly (mean age of 80 years) and rarely in young adults and children. As in most autoimmune diseases, there is a genetic component and the presence of human leukocyte antigen (HLA) associations support this (Petronic-Rosic, 2019). BP can also be associated with neurological diseases, such as multiple sclerosis, dementia, Parkinson Disease, cerebrovascular disease and epilepsy, which may predate the onset of BP by 5 years (Miyamoto et al., 2019). This association may be the result of a shared target antigen present in the skin and the central nervous system.
Certain drugs can trigger BP such as gliptins (vildagliptin, linagliptin), diuretics such as frusemide or spironolactone, and neuroleptics. The disease can occur within 3 months of starting the new medication and can reverse when the culprit drug is withdrawn (Miyamoto et al., 2019). There is no conclusive evidence for an association with malignancy.
What are the signs and symptoms of bullous pemphigoid?
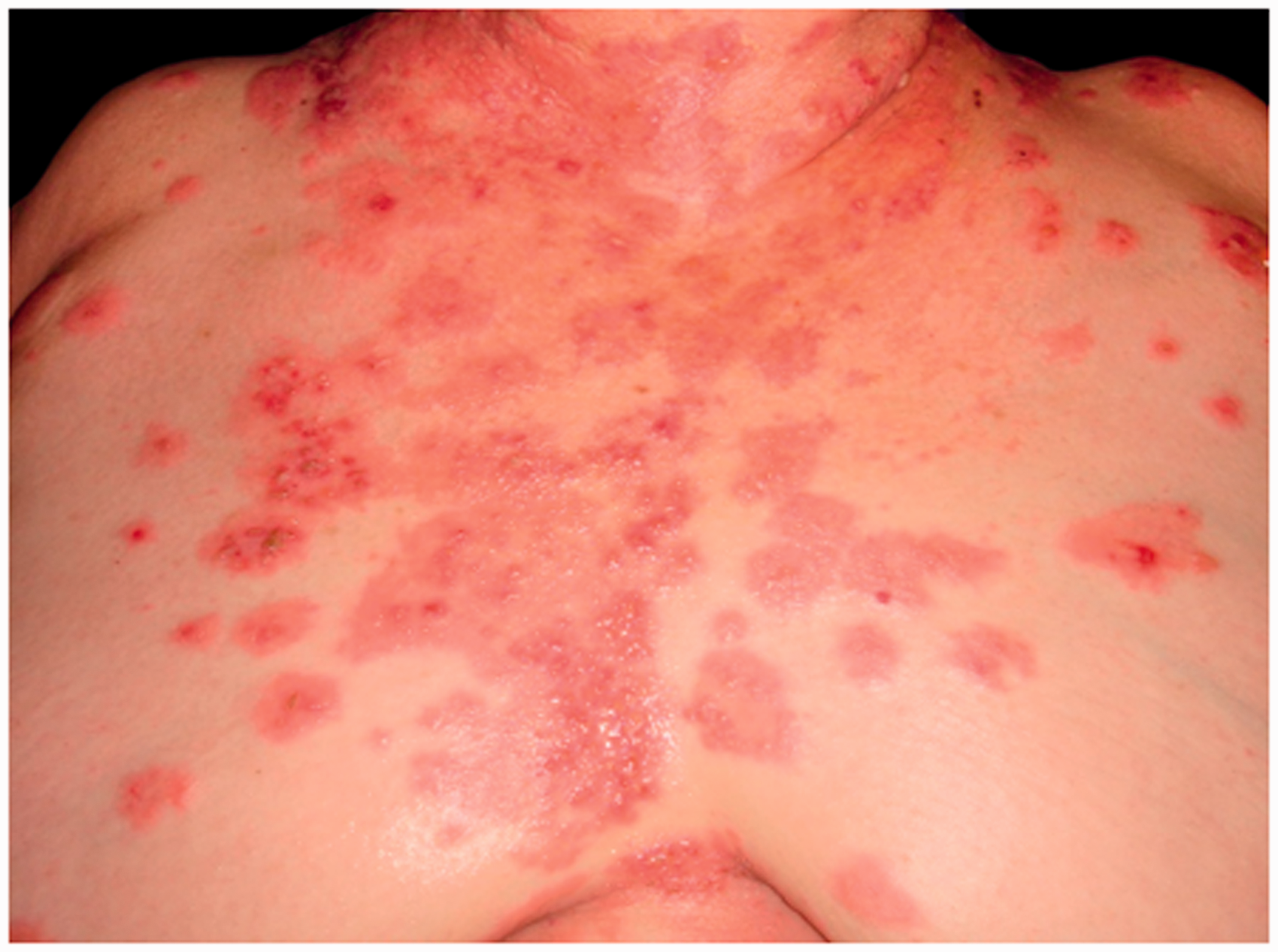
Classic BP can present with intense pruritus followed by the development of tense bullae (fluid-filled blisters), typically on the trunk and extremities (Figure 1). Some patients may present with early blistering on urticated plaques (Figure 2). Mucous membrane pemphigoid is a rarer subtype that can present with primarily mucosal involvement but can affect the skin and eyes (Oakley, 2016). Patients can present with oral blisters, sore eyes, nasal symptoms, change in voice, dysphagia and genital ulcers.
Classic tense blisters on an urticated background in bullous pemphigoid. Early blistering on urticated plaques.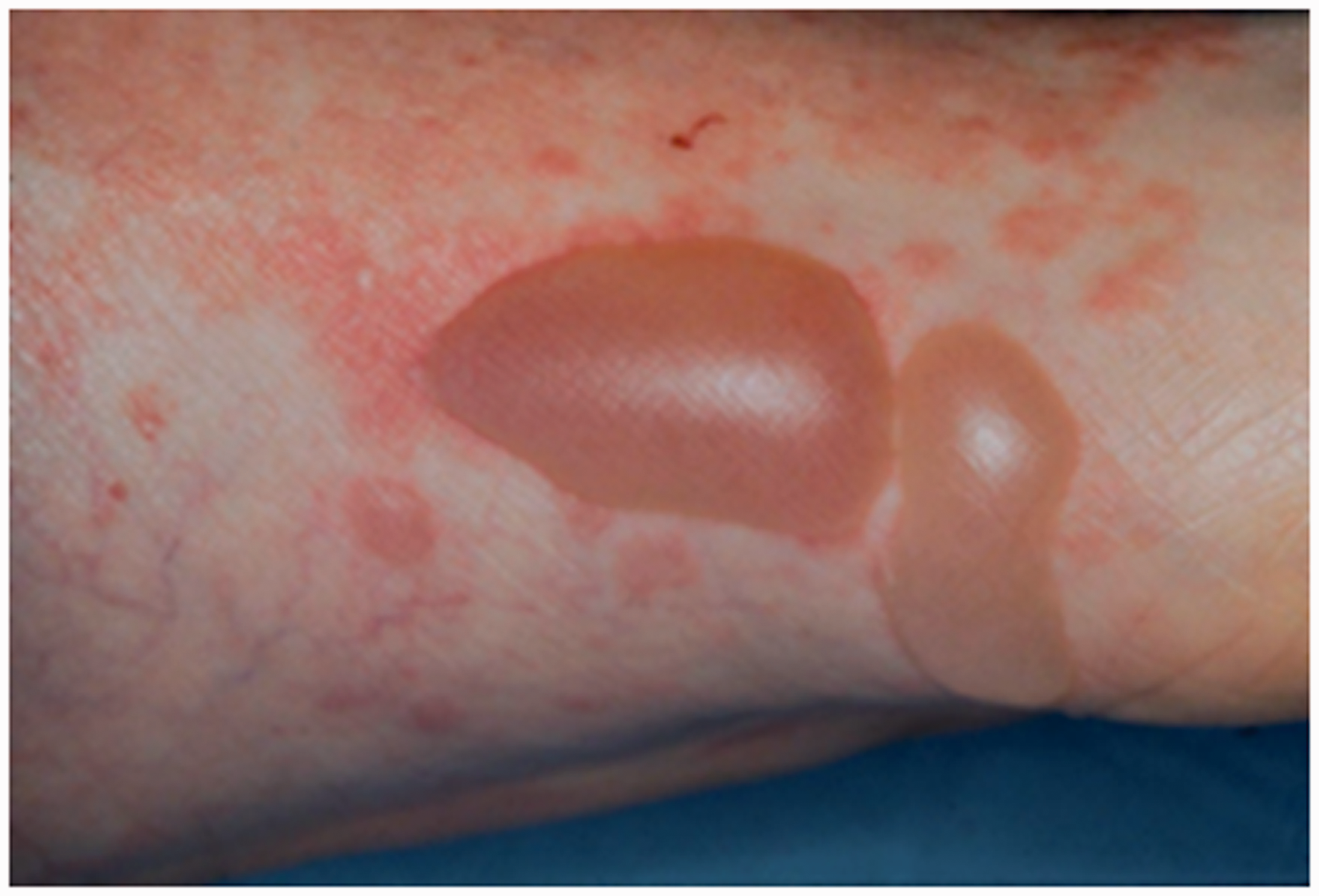
Lichen planus pemphigoides (LPP) has traditionally been thought to be a variant of BP, but recent studies suggest it may be a separate disease altogether (Hubner et al., 2019). These patients tend to first develop pruritic violaceus plaques and papules (lichen planus-like), which later blister (Figure 3). They may also have white lace-like lesions on the buccal mucosa. LPP responds to oral prednisolone and dapsone.
Violaceous papules, erosions and denuded blisters of lichen planus pemphigoides.
How is bullous pemphigoid diagnosed?
BP should be suspected in any elderly patient who presents with intense pruritus with or without any blistering, eczematous or urticarial rash (Oakley, 2016). Most patients with BP have raised serum eosinophils and therefore this can be a non-specific clue to the diagnosis (Lin et al., 2018). Once the blisters develop, the diagnosis is made clinically, and confirmed via immunofluorescence and histological analysis in secondary care. Blood tests are taken for indirect immunofluorescence, which measure circulating pemphigoid antibodies. A skin biopsy is taken for histology and direct immunofluorescence (DIF). Histological examination of the skin demonstrates the sub-epidermal split (blister) and presence of inflammatory cells, particularly eosinophils. DIF highlights the presence of tissue bound IgG antibodies and C3 deposition at the basement membrane.
The differential diagnosis for blistering eruptions is wide and includes bullous insect bites, drug eruptions, bullous cellulitis, erythema multiforme, porphyria and other autoimmune blistering diseases, such as epidermolysis bullosa acquisita.
What is the management of bullous pemphigoid?
The aim of treatment is to arrest formation of new lesions, promote healing and control pruritus. As the disease typically effects the elderly, the choice of treatment should take into account the patient’s ability to self-care and monitor for complications in order to reduce morbidity.
First-line treatment for BP is a potent topical corticosteroid (clobetasol propionate 0.05%) applied to the affected skin once a day (Venning et al., 2012). Oral prednisolone is often needed if topical application is not feasible for the patient at a dose of 0.5–1.0 mg/kg/day. Both topical and oral steroids are tapered down once symptoms are controlled. If steroids are not tolerated, contraindicated or the disease is relapsing, then high dose doxycycline (100 mg twice daily) can be given (Chalmers et al., 2017). In severe cases, additional medications, such as dapsone, methotrexate, azathioprine and mycophenolate mofetil, can be added if prednisolone alone is not controlling the disease. Intravenous immunoglobulin, rituximab or omilizumab are further options for treatment of refractory cases, but these are expensive and invasive (Miyamoto et al., 2019).
Wound care and nursing input are important. Blisters should generally be left intact unless they are large, tense and painful, which can then be pierced with a sterile needle. The roof of the blister is left to act as a natural dressing. Sometimes non-adherent dressings can be used for denuded blister sites.
BP is a chronic disease and primary care physicians can optimise care by monitoring for side effects of treatment and secondary infections. Referral to other specialities such as gastroenterology, gynaecology or ear, nose and throat may be appropriate to evaluate mucosal involvement. Hospital admission may be appropriate if there is widespread blistering or severe mucosal involvement (Rossi, 2018).