
Abstract
Background:
Although combinations of third-generation epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) with chemotherapy or amivantamab have significantly improved progression-free survival (PFS) and overall survival (OS) compared with EGFR-TKI monotherapy in EGFR-mutated advanced non-small cell lung cancer (NSCLC), the increased toxicities and requirement for intravenous administration of chemotherapy limit their practicality. Patients with EGFR-mutated advanced NSCLC who develop M1c2 disease or exhibit elevated programmed death-ligand 1 (PD-L1) expression represent a clinically refractory subgroup with a high unmet need for effective and convenient combination strategies.
Objectives:
This study is designed to evaluate the efficacy and safety of an all-oral regimen combining vinorelbine and firmonertinib (a third-generation EGFR-TKI) as first-line treatment in this refractory population.
Design:
This open-label, non-randomized, two-cohort study is conducted at two centers in China. Systematic treatment-naïve patients with histopathology confirmed EGFR-mutated (exon 19 deletion or exon 21 L858R mutation) NSCLC, who are not amenable to curative surgery or radiotherapy, will be recruited. Cohort 1 will comprise 54 metastatic NSCLC patients with M1c2 disease, and Cohort 2 will consist of 77 locally advanced or metastatic NSCLC patients with a PD-L1 tumor proportion score of ⩾25%. Recruitment began on December 11, 2025, and is currently in progress.
Methods and analysis:
During the one-cycle safety run-in period, six patients received oral vinorelbine (60 mg/m2 on days 1 and 8) plus firmonertinib (80 mg once daily) as the starting dose. Dosing regimen for the subsequent expansion cohort will be determined based on the safety data from the safety run-in period. The treatment cycle is repeated every 3 weeks and will continue until disease progression or unacceptable toxicity. The primary endpoint is investigator-assessed PFS. Secondary endpoints are objective response rate, disease control rate, OS, and safety.
Ethics:
This study has received ethics approval from the ethical committee of the Shanghai Chest Hospital (approval number: IS25204).
Discussion:
To the best of our knowledge, this study represents the first evaluation of vinorelbine plus firmonertinib in EGFR-mutated locally advanced or metastatic NSCLC with M1c2 disease or elevated PD-L1 expression, aiming to provide an effective, tolerable, and convenient all-oral treatment option for this refractory population.
Trial registration:
The study protocol (version 1.2; July 18, 2025) was registered on October 24, 2025, at the Chinese Clinical Trial Registry (ChiCTR2500111096).
Background
Epidermal growth factor receptor (EGFR) is a common driver oncogene in non-small cell lung cancer (NSCLC), with a prevalence of approximately 20% in White populations and 35% in Asian populations. 1 Approximately 85%–90% of EGFR alterations involve deletions in exon 19 (exon 19 deletion) and a leucine-to-arginine substitution in exon 21 (exon 21 L858R mutation). 2 For patients with EGFR-mutated locally advanced or metastatic NSCLC, the recommended first-line treatment option is third-generation EGFR-tyrosine kinase inhibitors (TKIs), such as osimertinib, aumolertinib, befotertinib, and firmonertinib.3–6 Despite high initial response rates, acquired resistance inevitably develops after a median progression-free survival (PFS) of 19–22 months, posing a major challenge to long-term disease control.3–8 To overcome or delay resistance and enhance efficacy, combination strategies incorporating third-generation EGFR-TKIs with chemotherapy or the EGFR-mesenchymal-epithelial transition factor bispecific antibody amivantamab have been explored, demonstrating significant improvements in median PFS and overall survival (OS) compared with EGFR-TKI monotherapy.9–12 However, these combination regimens are associated with additional and substantial toxicities, with grade ⩾3 adverse events (AEs) reported in approximately 70%–80% of patients.10,12 Notably, an early intersection of the OS curves between the combination and monotherapy groups was observed, suggesting that increased toxicities in some patients receiving combination therapy may have translated into a higher risk of all-cause mortality.10,12 Moreover, the requirement for intravenous administration of chemotherapy further limits treatment convenience. Besides, the phase III COMPEL trial showed that osimertinib plus chemotherapy was associated with improved PFS and OS compared with placebo plus chemotherapy in patients with EGFR-mutated advanced NSCLC following non-central nervous system progression on first-line osimertinib. 13 These findings support the feasibility of EGFR-TKI-based combination strategies but also point to the need for optimizing patient selection and the development of more tolerable and convenient therapeutic strategies.
Programmed death-ligand 1 (PD-L1), a widely used biomarker for predicting immunotherapy efficacy in NSCLC, has also attracted increasing attention for its impact on the efficacy and resistance to EGFR-TKIs.14,15 Multiple studies have shown that patients with positive (tumor proportion score (TPS) ⩾1%) PD-L1 expression treated with EGFR-TKIs exhibit significantly lower objective response rates (ORR) and shorter median PFS than those with negative (TPS <1%) PD-L1 expression, and thus are more likely to develop primary resistance to EGFR-TKIs.16–20 The phenomenon still exists when the cutoff value of TPS is changed to 25%.20,21 Meanwhile, analyses based on the ninth edition of the tumor, node, and metastasis (TNM) database have revealed that a greater number of metastatic lesions is associated with poorer prognosis.22,23 Among patients with multiple metastases, survival outcomes are particularly unfavorable when multiple organ systems are involved (M1c2). 23 Taken together, EGFR-mutated advanced or metastatic NSCLC with elevated PD-L1 expression or M1c2 disease represents a clinically challenging and refractory subgroup. In this context, third-generation EGFR-TKI monotherapy may be insufficient for durable disease control, and combination treatment strategies may therefore be required.
Firmonertinib (AST2818; formerly furmonertinib) is a novel, oral, highly brain-penetrant third-generation EGFR-TKI that broadly and selectively targets EGFR mutations. 24 It has demonstrated promising efficacy with an acceptable safety profile in EGFR-mutated locally advanced or metastatic NSCLC, including patients with central nervous system metastases.5,25,26 Based on these pivotal clinical data, firmonertinib has been approved by the National Medical Products Administration (NMPA) of China for the treatment of locally advanced or metastatic NSCLC patients harboring acquired EGFR T790M mutation, and as first-line treatment for patients with EGFR exon 19 deletion or exon 21 L858R mutations.5,25,27 Vinorelbine, a semi-synthetic vinca alkaloid that inhibits microtubule assembly, is an established cytotoxic agent for NSCLC, either as monotherapy or as part of combination chemotherapy.28,29 Preclinical studies have demonstrated that vinorelbine combined with EGFR-TKIs exerts synergistic anti-tumor effects in NSCLC cells, irrespective of EGFR mutation status.30–32 Importantly, vinorelbine is also available in an oral formulation, improving treatment convenience. In light of these findings, we have designed this multicenter, non-randomized, two-cohort study of vinorelbine plus firmonertinib, with the aim of providing an effective, safe, and convenient all-oral treatment option for EGFR-mutated advanced or metastatic NSCLC with M1c2 disease or elevated PD-L1 expression (TPS ⩾25%).
Materials and methods
Study design
This is a multicenter, open-label, non-randomized, two-cohort study evaluating the efficacy and safety of a first-line, all-oral regimen of vinorelbine plus firmonertinib in refractory (M1c2 stage or elevated PD-L1 expression) EGFR-mutated NSCLC. Recruitment began on December 11, 2025, and is currently in progress. Eligible patients will be recruited from two centers (Shanghai Chest Hospital and Shanghai GoBroad Cancer Hospital) in China. The study design is shown in Figure 1. Cohort 1 will include patients with EGFR-mutated metastatic NSCLC who meet the M1c2 category (involvement of multiple extrathoracic organ systems) according to the ninth edition of the TNM classification for lung cancer.22,23 Cohort 2 will include patients with EGFR-mutated locally advanced or metastatic NSCLC and elevated PD-L1 expression (TPS ⩾25%). For patients who exhibit both M1c2 disease and elevated PD-L1 expression, cohort assignment will be determined by the principal investigator or clinical physicians based on patients’ specific clinical characteristics; no patient will be enrolled in both cohorts simultaneously. The reporting of this study conforms to SPIRIT statement (see Supplemental Appendix 1). 33
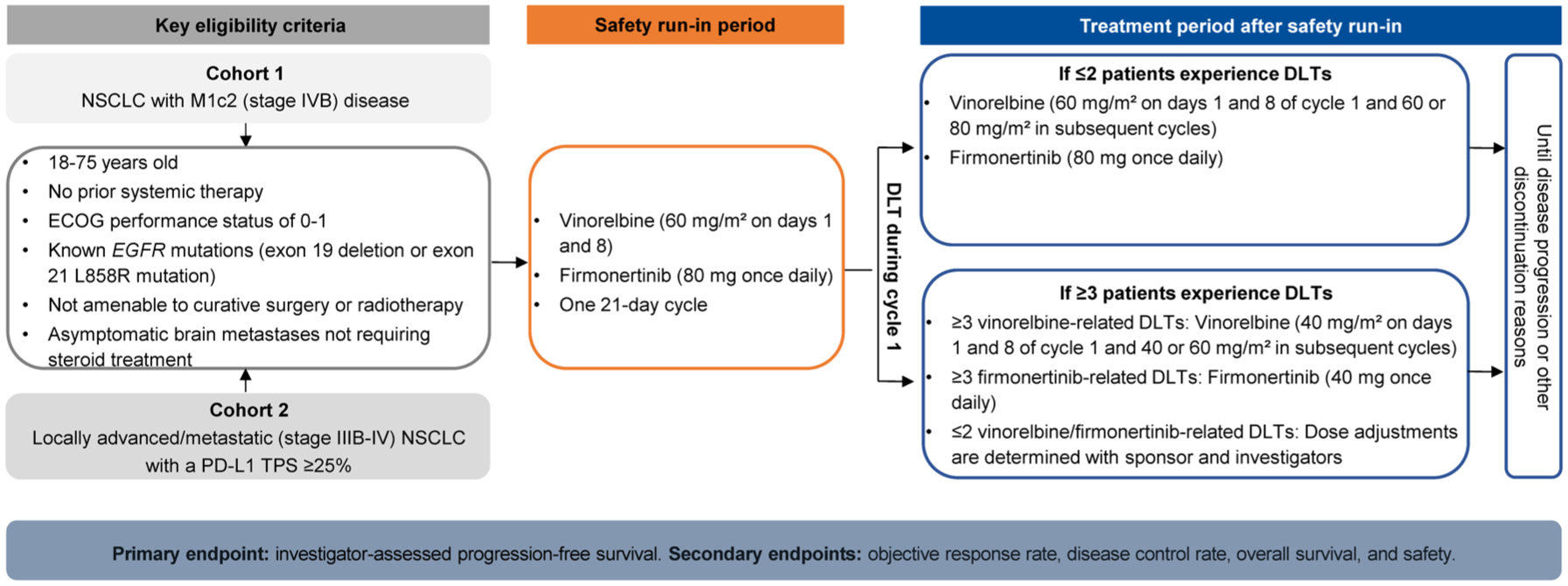
Study design.
Eligibility criteria
Eligible patients are aged 18–75 years with histologically confirmed EGFR-mutated (exon 19 deletion or exon 21 L858R mutation) locally advanced or metastatic NSCLC, who are not amenable to curative surgery or radiotherapy. Detailed inclusion and exclusion criteria are listed in Table 1. Patients may enter the trial and initiate the planned treatment only after eligibility has been confirmed by the investigators and written informed consent has been obtained from the patients or their legally authorized representatives.
Detailed inclusion and exclusion criteria.
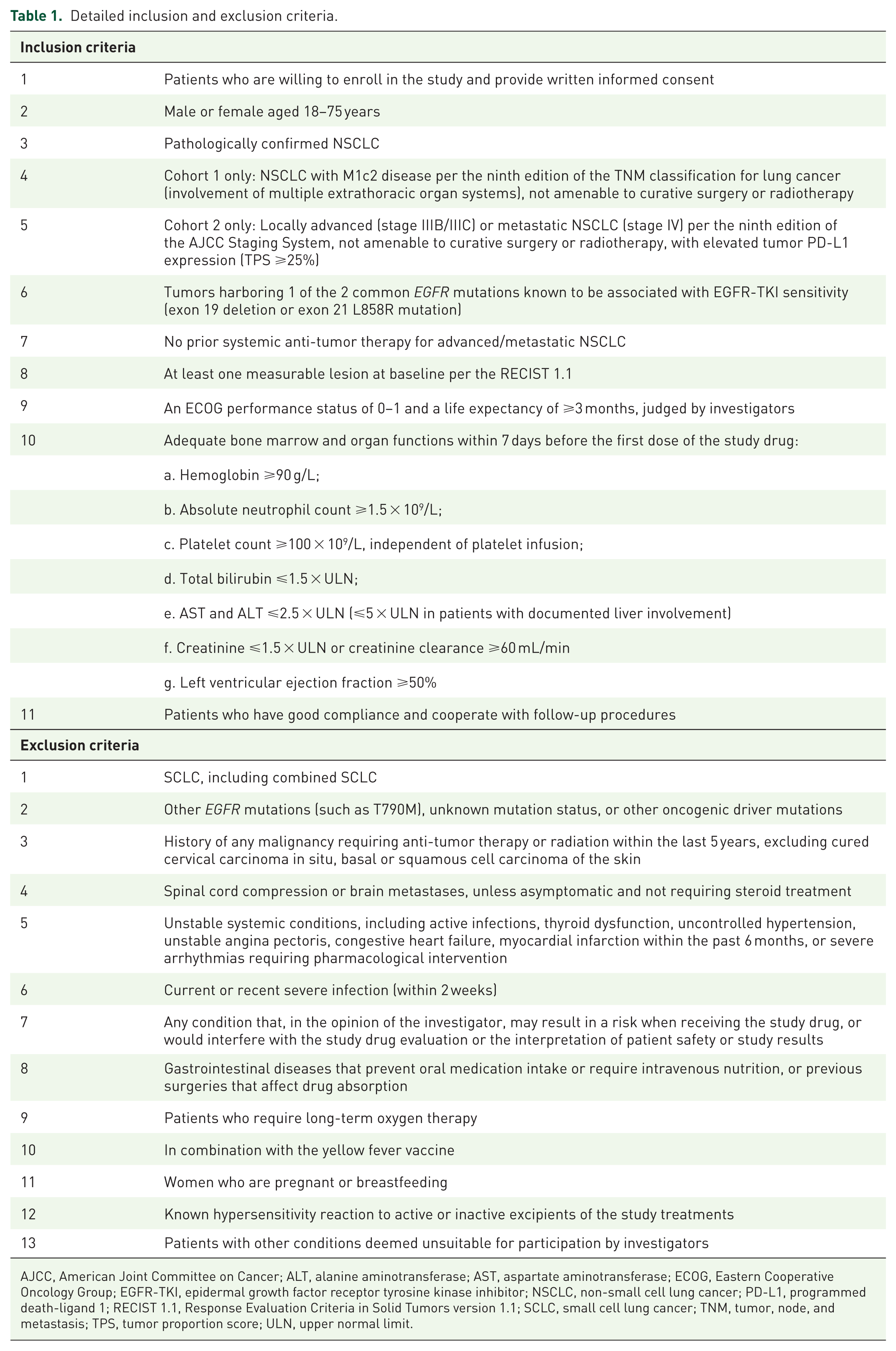
AJCC, American Joint Committee on Cancer; ALT, alanine aminotransferase; AST, aspartate aminotransferase; ECOG, Eastern Cooperative Oncology Group; EGFR-TKI, epidermal growth factor receptor tyrosine kinase inhibitor; NSCLC, non-small cell lung cancer; PD-L1, programmed death-ligand 1; RECIST 1.1, Response Evaluation Criteria in Solid Tumors version 1.1; SCLC, small cell lung cancer; TNM, tumor, node, and metastasis; TPS, tumor proportion score; ULN, upper normal limit.
Procedures
The study will begin with a safety run-in period, during which six patients from either cohort will receive oral vinorelbine (60 mg/m2 on days 1 and 8) plus firmonertinib (80 mg once daily) every 3 weeks. Safety and tolerability of this combination regimen at this dose level will be assessed based on dose-limiting toxicity (DLT) occurring during the first treatment cycle. Patients who do not complete the DLT assessment window (the first treatment cycle) for reasons unrelated to toxicity (e.g., early withdrawal) will be replaced to ensure adequate DLT evaluability. DLTs are defined as any of the following AEs related to the study drug from the first dose to the end of the first treatment cycle: (1) Grade 4 neutropenia lasting ⩾7 days; (2) Febrile neutropenia; (3) Grade 4 thrombocytopenia; (4) Grade 3 thrombocytopenia with severe bleeding; (5) Grade 4 anemia; (6) Grade ⩾3 non-hematologic toxicities lasting >7 days, excluding isolated, asymptomatic laboratory abnormalities. During the safety run-in period, if ⩽2 patients experience DLTs, the combination dose for this study will be established as vinorelbine 60 mg/m2 on days 1 and 8 of cycle 1, with subsequent cycles maintained at 60 mg/m2 or escalated to 80 mg/m2 based on hematologic toxicity, combined with firmonertinib 80 mg once daily. If ⩾3 patients experience DLTs, the sponsor and investigator will determine drug attribution and implement dose de-escalation as follows: (1) If ⩾3 DLTs are attributed to vinorelbine, the vinorelbine dose in cycle 1 will be reduced to 40 mg/m2, with subsequent cycles maintained at 40 mg/m- or escalated to 60 mg/m2 based on hematologic toxicity; (2) If ⩾3 DLTs are attributed to firmonertinib, the firmonertinib dose for the entire study will be reduced to 40 mg/day; (3) If ⩽2 DLTs are attributed to each drug, further dose adjustments will be determined through joint assessment with sponsor and investigators.
Patients will continue receiving the study treatment until disease progression, death, investigator decision, unacceptable toxicity, withdrawal of consent, or any other reasons for discontinuation as determined by the investigators. Dose reductions for vinorelbine (from 80 to 60 mg/m2 or from 60 to 40 mg/m2) or firmonertinib (from 80 to 40 mg), as well as temporary dose interruptions of up to 3 weeks, are permitted to manage treatment-related AEs. If AEs fail to resolve within 3 weeks, the corresponding drug will be permanently discontinued. Patients who discontinue vinorelbine due to unacceptable toxicity may continue firmonertinib, and vice versa.
From the time of signing the informed consent form until the end-of-treatment visit, patients are prohibited from receiving any anti-tumor therapies not specified in the protocol, including radiotherapy or local therapy confined to target lesions, surgery, chemotherapy, biological agents, or other investigational agents. Strong CYP3A4 inhibitors or inducers should be avoided during treatment with vinorelbine and firmonertinib. Medications and therapies for managing complications or alleviating their symptoms, such as antihypertensives, antibacterials, and antihistamines, are permitted.
Tumor assessments using computed tomography or magnetic resonance imaging are performed at baseline and every 6–8 weeks until disease progression or initiation of new anti-tumor therapy, whichever occurs first. Tumor responses are evaluated by investigators at each study site according to the Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST 1.1) and subsequently reviewed by the leading principal investigator at the primary center to ensure consistency. In addition, any discordant assessments will undergo repeat radiologic review for confirmation. Survival is assessed every 2 months following treatment discontinuation. Safety is evaluated by recording the incidence, severity, and causality of AEs occurring from the time of signing of the informed consent form until 30 days after the last dose of the study treatment. AEs are assessed at each visit through laboratory tests (such as hematology, urinalysis, and chemistry), vital signs, physical examinations, echocardiography, 12-lead electrocardiograms, and Eastern Cooperative Oncology Group performance status scores, and are graded according to the Common Terminology Criteria for Adverse Events version 5.0.
EGFR mutations are detected during screening using tumor tissue samples by either next-generation sequencing or amplification refractory mutation system method. Baseline PD-L1 expression is assessed in tumor tissue samples using the PD-L1 immunohistochemistry 22C3 pharmDx assay (Agilent Technologies, Carpinteria, CA, USA). PD-L1 expression is categorized by TPS, which is defined as the percentage of tumor cells exhibiting positive PD-L1 staining among all tumor cells.
Outcomes
The primary endpoint is PFS assessed by investigators per the RECIST 1.1, which is defined as the time interval from the first dose of study treatment until disease progression or death from any cause, whichever occurs first. The secondary endpoints include ORR (the sum of complete response (CR) and partial response (PR) at the best overall response), disease control rate (the sum of CR, PR, and stable disease (SD) at the best overall response), OS (time interval from the first dose of study treatment to death from any cause), and safety.
Statistical analysis
Sample sizes for Cohorts 1 and 2 are estimated based on the primary endpoint of PFS, using PASS software (version 15.0.5; NCSS, Kaysville, UT, USA). Based on available evidence from other EGFR-TKIs in clinically similar EGFR-mutated subgroups and our unpublished retrospective data, the median PFS with firmonertinib monotherapy is assumed to be 8.0 months for Cohort 1 and 9.0 months for Cohort 2.34,35 Assuming an exponential distribution for PFS, the combination regimen is expected to improve the median PFS to 13.3 months in Cohort 1 (hazard ratio (HR) = 0.60) and 13.8 months in Cohort 2 (HR = 0.65). Under these assumptions, 49 patients in Cohort 1 and 70 patients in Cohort 2 are deemed sufficient to achieve a significance level of 0.05 and a statistical power of 80%. Considering an anticipated 10% dropout rate, a total of 131 patients (54 in Cohort 1 and 77 in Cohort 2) are required.
Efficacy analyses will be performed on both the intention-to-treat (ITT) and per-protocol set (PPS) populations. The ITT population includes all enrolled patients, while the PPS population comprises all enrolled patients who receive at least one dose of the study treatment without major protocol violations. Major protocol violations include failure to meet the inclusion or exclusion criteria, concomitant use of prohibited medications during the study period that seriously affect the assessment of study endpoints, non-adherence to the study protocol, or other deviations that seriously impact the reliability of study endpoints. Safety will be assessed in all enrolled patients who receive at least one dose of the study treatment (safety analysis set population). Baseline characteristics and safety data are descriptively summarized with median (interquartile range), median (range), or frequency (percentage). The Kaplan–Meier method will be used to estimate the medians of time-to-event endpoints, and the Brookmeyer–Crowley method will be used to calculate corresponding 95% confidence intervals (CIs). For the primary endpoint of PFS, differences between the trial group (Cohort 1 or 2) and the corresponding historical control group (median PFS: 8 months for Cohort 1 and 9 months for Cohort 2) will be tested using the log-rank test. Response rates with corresponding 95% CIs will be calculated with the Clopper–Pearson method. All statistical analyses were performed using the SAS Software (version 9.4 or later; SAS Institute Inc., Cary, NC, USA), and statistical significance was set at a two-sided p < 0.05. The two cohorts will be analyzed independently, with no cross-cohort comparisons. As each cohort is based on an independent study objective and predefined statistical hypothesis, no multiplicity adjustment is planned. Besides, a pre-specified sensitivity analysis will be conducted for the primary endpoint in each cohort, excluding patients eligible for both cohorts, to assess the robustness of the primary findings to this potential source of variability.
Discussion
To overcome or delay resistance to third-generation EGFR-TKIs, combination strategies (such as EGFR-TKIs plus chemotherapy or amivantamab) have been explored to achieve durable disease control in patients with EGFR-mutated locally advanced or metastatic NSCLC.9–13 However, identifying the subset of patients most likely to benefit from such combinations remains a key challenge, as some may achieve favorable outcomes with EGFR-TKI monotherapy and thereby avoid the additional toxicity associated with combination regimens. Patients with M1c2 disease exhibit a worse prognosis, and elevated PD-L1 expression is associated with inferior treatment outcomes, representing clinically challenging and refractory subgroups with a greater need for effective combination strategies.16–20,23 To our knowledge, this is the first prospective trial evaluating the clinical benefits of vinorelbine plus firmonertinib as a first-line treatment in this selected population of patients with EGFR-mutated advanced or metastatic NSCLC with M1c2 disease or elevated PD-L1 expression (TPS ⩾25%).
The combination of vinorelbine plus firmonertinib may offer advantages in terms of safety and treatment convenience. Building on the established activity of vinorelbine monotherapy in advanced NSCLC, this study adopts a strategy of single-agent chemotherapy combined with a third-generation EGFR-TKI.28,29 Compared with the platinum-pemetrexed doublet chemotherapy used in the phase III FLAURA2 trial, single-agent chemotherapy with vinorelbine is theoretically associated with a lower toxicity burden.9,10 Additionally, in contrast to the intravenous chemotherapy used in the FLAURA2 trial, vinorelbine can be administered orally.9,10 The totally intravenous-free regimen of vinorelbine plus firmonertinib allows for at-home self-administration, thereby reducing the need for venous access and the risk of catheter-related infections, while also minimizing frequent outpatient visits and healthcare resource utilization.36,37 Collectively, these features may contribute to improved health-related quality of life, which is especially meaningful in the long-term management of EGFR-mutated advanced or metastatic NSCLC.
The key limitation of this study was its open-label, which may introduce bias in investigator-assessed outcomes. Nevertheless, to minimize potential assessment bias, response assessments were rechecked by the leading principal investigator at the primary center after evaluations by investigators at each study site. Besides, the non-randomized design precludes direct comparisons of the efficacy and safety of the combination regimen with EGFR-TKI monotherapy or other combination strategies. The use of historical controls derived from other EGFR-TKIs may also limit comparability owing to the lack of firmonertinib-specific data in these subpopulations. Although the study is designed with a predefined statistical hypothesis and sample size calculation, the relatively small sample sizes in the two cohorts warrant cautious interpretation of the results. Moreover, the study population consists exclusively of Chinese patients, and firmonertinib is currently approved only by the NMPA in China, which may limit the generalizability and global applicability of these findings.
Conclusion
Based on the established anti-tumor activity of vinorelbine and firmonertinib as single agents, together with their potential synergistic effects, this all-oral combination of a third-generation EGFR-TKI and single-agent chemotherapy represents an attractive therapeutic strategy. The results of this study will help define the clinical role of vinorelbine plus firmonertinib in EGFR-mutated locally advanced or metastatic NSCLC with M1c2 disease or elevated PD-L1 expression (TPS ⩾25%), with the goal of delaying EGFR-TKI resistance while reducing toxicity and improving treatment convenience.
Supplemental Material
sj-docx-1-tam-10.1177_17588359261455295 – Supplemental material for Efficacy and safety of first-line oral vinorelbine plus firmonertinib in refractory EGFR-mutated non-small cell lung cancer: study protocol of a multicenter, non-randomized, two-cohort study
Supplemental material, sj-docx-1-tam-10.1177_17588359261455295 for Efficacy and safety of first-line oral vinorelbine plus firmonertinib in refractory EGFR-mutated non-small cell lung cancer: study protocol of a multicenter, non-randomized, two-cohort study by Bo Zhang, Yanwei Zhang, Chunlei Shi, Zhiqiang Gao, Runbo Zhong, Rong Qiao, Shuang Li, Hua Zhong and Baohui Han in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
This study was supported by the Clinical Research Special Fund Project of Shanghai Chest Hospital (project number: 2024IIT-Q004). This study was supported by Pierre Fabre (Shanghai) Medical Co., Ltd., with special thanks to Ziyu Guo, Zhuoqiong Ren, and Fang Su from its Medicine Department, as well as Shanghai Allist Pharmaceuticals. The authors thank all patients, their families, investigators, and research staff for participating in this trial.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.