
Abstract
Cytokine release syndrome (CRS) represents a spectrum of immune hyperactivation initiated by rapid T-cell engagement and amplified through innate immune cells, particularly monocytes and macrophages, with downstream cytokine cascades driving endothelial dysfunction, capillary leak and organ impairment. CRS is a frequent, class-associated toxicity of T-cell-engaging immunotherapies and is being observed with increasing regularity as T-cell-redirecting therapies progress through clinical development in solid tumours. Although CRS is typically less severe in patients with solid tumours relative to those with haematological malignancies, phase II–III trials of these emerging agents consistently report grade 1–2 CRS in the majority of patients, highlighting the need for effective prevention and management strategies, while preserving anti-tumour activity of the primary therapy. Current grading and management algorithms have been largely extrapolated from experience with chimeric antigen receptor T-cell therapies and rely on step-up dosing, premedication, supportive care and escalation to interleukin (IL)-6/IL-6R blockade and corticosteroids. However, steroid-refractory or high-grade CRS, as well as overlapping syndromes underscore the limitations of this approach and the need for mechanism-based interventions targeting upstream mediators and signalling hubs. These include IL-1 blockade, Janus kinase-signal transducer and activator of transcription inhibition, granulocyte–macrophage colony-stimulating factor neutralisation, tumour necrosis factor-alpha inhibition and interferon gamma blockade. In this narrative review, we synthesise the mechanistic rationale and emerging clinical evidence underpinning CRS prevention and management in solid tumours, with a particular focus on T-cell engager (TCE) and other T-cell-redirecting therapies. We then outline key risk-mitigation strategies and discuss their potential implications for anti-tumour immunity. Finally, we highlight key knowledge gaps, particularly the paucity of prospective solid tumour-specific data, with current CRS prevention and management strategies therefore remaining largely extrapolated from haematological malignancies, alongside the need for biomarker-driven risk stratification, optimal sequencing and prophylactic strategies and the development of next-generation TCE designs.
Keywords
Introduction
Over the past decade, immune checkpoint inhibitors (ICIs) have transformed the therapeutic landscape in oncology, with durable survival benefits observed in multiple solid tumours, such as melanoma, lung cancer and renal cell carcinoma.1–3 As such, novel immunotherapeutic agents continue to be developed, with some demonstrating promising clinical activity. 4 Adoptive cell therapies, including tumour-infiltrating lymphocyte (TIL) therapy, are now being evaluated in phase III trials in solid tumours, while T-cell engagers (TCEs), such as tebentafusp and tarlatamab, have already been adopted as standard of care in uveal melanoma and are reshaping second-line in small-cell lung cancer, although approval status and access remain geographically variable.4–8
TCEs are molecules engineered to redirect cytotoxic T cells against tumour cells by simultaneously binding CD3 on T cells and a tumour-associated antigen, thereby mediating major histocompatibility complex-independent tumour cell lysis. 9
Besides these respective binding domains, TCEs are designed with either an active or Fc-silent domain. 10 TCEs with an active Fc domain can also mediate antibody-dependent and complement-dependent cytotoxicity, whereas Fc-silent agents are typically associated with reduced off-tumour toxicity and cytokine release syndrome (CRS). 11
Irrespective of the scaffold, T-cell activation remains intrinsically linked to a characteristic toxicity profile driven by immune hyperactivation and CRS. 12 In the context of TCEs, phase II–III trials in solid tumours have reported CRS in approximately 50%–80% of patients, albeit predominantly grades 1–2, underscoring that CRS remains a frequent on-target toxicity in this setting.5,6 Importantly, CRS can manifest with a broad clinical spectrum, ranging from mild, self-limiting episodes to fulminant, life-threatening events. 13 Patients typically present with high fever initially, and can rapidly progress to hypotension, hypoxia and multi-organ dysfunction. 13 Prompt identification and intervention are essential to enable accurate diagnosis, guide management decisions and ensure timely escalation of care, particularly to distinguish CRS from sepsis, tumour-related fever and other inflammatory syndromes.
In this narrative review, we summarise the current understanding of the biological mechanisms driving CRS induced by TCEs and other T-cell-redirecting therapies in solid tumours. We then discuss emerging mechanism-based therapeutic strategies targeting key cytokines and signalling pathways, with the aim of optimising CRS management while preserving anti-tumour immunity and enabling a more precise and rational use of immunotherapy in this setting.
Overview of CRS in solid tumours across T-cell-redirecting therapies
Definition and grading
The American Society for Transplantation and Cellular Therapy (ASTCT) provides the most widely accepted consensus grading system for CRS. 14 In this consensus, severity is defined primarily by the degree of hypoxia and hypotension, with the presence of fever constituting an essential diagnostic criterion. 14 Comparable grading systems have been proposed, including CTCAE v5.0, the Lee criteria, Penn criteria, MSKCC and CARTOX.15–18 Across systems, grade 1 CRS is characterised by mild symptoms without evidence of organ dysfunction, while grade 2 CRS reflects the need for moderate supportive measures such as intravenous fluids or supplemental oxygen. Grades 3 and 4 CRS correspond to severe or life-threatening organ dysfunction, respectively, requiring escalating interventions such as vasopressors, high-flow oxygen or mechanical ventilation.
Some criteria also stipulate timeframes for intervention, specify the type of supportive measures required or include threshold levels for oxygen supplementation, often using arbitrary cut points. 18 Elevated serum cytokines, including interleukin (IL)-6, interferon gamma (IFN-γ) and IL-10, have been incorporated into certain grading systems but are generally considered impractical for clinical use.17,19 Although these grading systems were developed in the context of chimeric antigen receptor (CAR) T-cell therapy, they have subsequently been extrapolated to CRS arising from other treatment modalities, including TCEs and TIL therapy.
Incidence
Variability in the definitions and grading criteria for CRS, as outlined before, complicates comparisons across therapeutic platforms and may further contribute to underreporting. 20 Reported rates of CRS are typically highest with CAR-T cell therapies, lower with TCEs and uncommon with ICIs and TIL therapy.21,22 Severity also varies substantially between immune-stimulating agents. A comparative analysis reported grade 3 CRS in 4.9% of patients treated with bispecific TCEs, compared with 46% among those receiving CAR-T therapy. 23 Other studies have described similar or lower rates of severe CRS with TCEs, reflecting differences in dosing schedules and mitigation strategies such as corticosteroid premedication or step-up dosing.13,19,23
A recent systematic review and meta-analysis, including studies published up to June 2025, identified 15 TCEs evaluated in solid tumours. 20 Among 2439 treated patients, grade 1–2 inflammatory disorders occurred in 60.2%, while grade ⩾3 events were observed in 5.3%. The term ‘inflammatory disorder’ was used to encompass both confirmed CRS and CRS-like symptom clusters, acknowledging that true CRS incidence may be under-reported due to symptom overlap with other toxicities. Incidence estimates for adoptive cell therapies in solid tumours are limited by the early termination and small size of many studies.
In addition, we conducted a systematic search of PubMed and ClinicalTrials.gov (Supplemental Appendix 1) and identified 20 eligible TCR-T and CAR-T trials in solid tumours, including a total of 543 patients. Safety outcomes were extracted and pooled, yielding CRS rates of 56.5% for grades 1–2 and 3.7% for grades 3–5 toxicity (Table 1). However, heterogeneity in grading systems limits comparability and precludes formal meta-analytic weighting. Furthermore, TIL therapy poses an additional diagnostic challenge, as toxicities from non-cellular components such as high-dose IL-2 can mimic or mask CRS. Severe CRS is rare, though subclinical or CRS-like presentations may occur, characterised by pyrexia and hypotension. 24 A pooled analysis of 40 TIL trials reported pyrexia of any grade in 56.1% of patients and grade ⩾3 pyrexia in 24.8%. 25
CRS incidence in adoptive cellular therapy trials for solid tumours (TCR-T and CAR-based trials).
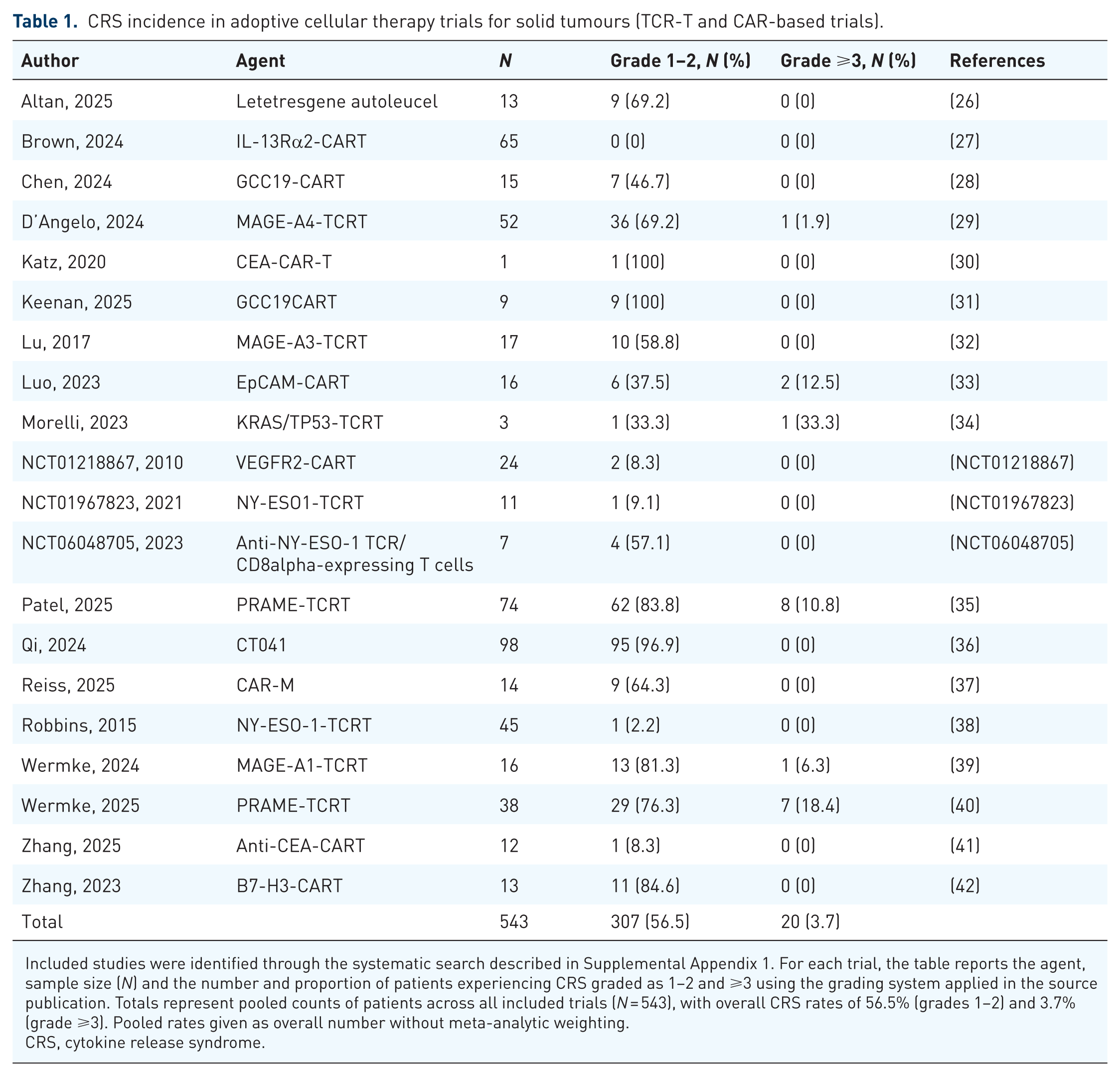
Included studies were identified through the systematic search described in Supplemental Appendix 1. For each trial, the table reports the agent, sample size (N) and the number and proportion of patients experiencing CRS graded as 1–2 and ⩾3 using the grading system applied in the source publication. Totals represent pooled counts of patients across all included trials (N = 543), with overall CRS rates of 56.5% (grades 1–2) and 3.7% (grade ⩾3). Pooled rates given as overall number without meta-analytic weighting.
CRS, cytokine release syndrome.
Clinical presentation
CRS typically presents with a constellation of systemic inflammatory symptoms (Table 2). Given the non-specific nature of many symptoms, alternative aetiologies must be excluded; in this context, careful attention to the temporal relationship between treatment and symptom onset is crucial for establishing causality. CRS associated with immune-effector cell engagement typically occurs within the first 14 days of treatment, often peaking within days of administration, corresponding to maximal in vivo T-cell expansion.14,15 With TCEs such as tarlatamab, CRS onset is usually rapid, with a median time to onset of 13.7 h and a median resolution time of 3 days. 43 In contrast, ICI-associated CRS is frequently delayed: one case series reported a median time to fever onset of 11 days following ICI initiation, while an analysis of the WHO pharmacovigilance database identified cases presenting up to 4 weeks post-treatment.22,44
Differential diagnosis of CRS and CRS-like presentations in patients receiving immune-stimulating therapies.

Uninterpretable in the context of anti-IL6R use.
CRP, C-reactive protein; CRS, cytokine release syndrome; HLH, haemophagocytic lymphohistiocytosis; IRR, infusion-related reaction; MAS, macrophage activation syndrome; SIRS, systemic inflammatory response syndrome.
The haemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS) spectrum overlaps substantially with CRS. HLH is characterised by high fever, hyperferritinaemia and cytopenias, and is often life-threatening. 45 It is a recognised complication of CAR-T and CAR-NK therapies and has been increasingly reported, albeit rarely, with ICIs.16,45 CRS-like presentations may also be attributable to concomitant therapies within multi-modal treatment regimens. Lymphodepletion preceding adoptive cell therapy increases the risk of febrile neutropenia and sepsis with associated systemic inflammatory response syndrome. By contrast, TCEs do not require lymphodepleting preconditioning.19,27
Risk factors
In haematological malignancies, recognised risk factors for CRS include higher doses of the causative agent, greater tumour burden and inter-current infection.15,19 Several of these factors also predispose to other complications such as tumour lysis syndrome. 16 Rising C-reactive protein (CRP) levels have been shown to predict severe CRS, while elevated concentrations of multiple cytokines within 36 h of CAR-T infusion similarly correlated with grade ⩾3 events.55,56 As CRS arises from antigen engagement by activated T cells, greater target density and binding efficiency are linked to more severe manifestations. This may explain the relatively lower incidence of severe CRS with CAR-T constructs in solid tumours, where effective trafficking and antigen engagement are hindered by the tumour microenvironment. 57 Similar principles extend to other CRS-associated agents in solid tumours, although small molecule inhibitors may achieve higher intratumoural penetration than immune effector cells. 58
Dosing strategy may further modulate CRS risk. Subcutaneous administration is being explored as a means of attenuating systemic cytokine peaks and reducing CRS incidence. 56 Conversely, the use of granulocyte colony-stimulating factor (G-CSF) following lymphodepleting chemotherapy has been associated with an increased risk of severe CRS. 59 Accordingly, G-CSF is generally avoided during the high-risk period except in contexts such as TIL therapy, where CRS risk is minimal. 21
In solid tumours treated with T-cell-redirecting therapies, the incidence and severity of CRS has been shown to correlate with dose intensity and typically occurs after the first administrations of the drug. 60 While some evidence suggests a higher risk of CRS in patients with greater tumour burden, this association remains uncertain, as other analyses have not demonstrated a clear correlation between baseline ctDNA levels and the incidence of CRS. 61
Immunopathogenesis
Although the mechanisms by which CRS occur are poorly defined, these are thought to occur as a result of ‘on-target’ effects leading to endogenous immune activation triggered by effector immune cell engagement. Upon recognition of tumour antigens, CD4+ and CD8+ effector T cells, NK cells and myeloid cells secrete pro-inflammatory cytokines that recruit and activate additional immune cell subsets. 15 The inflammatory process is further amplified through IL-6 and IL-1 secretion by activated macrophages and monocytes. 62 Whereas tumour necrosis factor-alpha (TNF-α), granulocyte–macrophage CSF (GM-CSF) and IFN-γ are responsible for initial immune system activation, IL-6, IL-8 and IL-18 amplify responses leading to further systemic inflammation and vascular permeability. 63
This excessive cytokine milieu leads to endothelial activation, coagulopathy and immune effector cell-associated neurotoxicity syndrome, accounting for the clinical symptoms of CRS. The magnitude of CRS can be intensified in the presence of increased tumour antigen load, enhancing immune effector cell engagement and subsequent cytokine release, whereas dense stromal networks and restricted vasculature limit immune infiltration and cytokine diffusion. 64 Together, these mechanisms highlight how immune cell activation, cytokine signalling and tumour microenvironment dynamics converge to determine CRS severity and outcome and provide the basis for the mechanism-based therapeutic strategies outlined in Figure 1.

Mechanistic overview of CRS associated with CD3-directed TCEs and key therapeutic management.
Mechanism-based therapeutic armamentarium
IL-6/IL-6R blockade
IL-6 is a key downstream mediator of CRS, contributing to fever, capillary leak and vascular permeability, hypotension and acute-phase responses. Accordingly, IL-6 pathway blockade is a central mechanism-based intervention and is widely used across immune effector therapies. Tocilizumab (anti-IL-6R) is the most established option, with regulatory approval for severe or life-threatening CAR-T cell-induced CRS and extensive real-world experience, typically leading to rapid improvement in fever and haemodynamics. 65 This approach has been extrapolated to TCEs and is commonly recommended for persistent grade 2 or higher CRS, alongside supportive care and escalation to corticosteroids for severe or refractory cases. In practice, tocilizumab is usually given as 8 mg/kg intravenously (maximum 800 mg) over 1 h, with repeat dosing if clinically indicated (typically at 6–8 h intervals maximum of 3 doses in 24 h and 4 doses total per CRS episode), integrated within local CRS pathways. 66 Siltuximab (anti-IL-6) is an alternative strategy that binds and neutralises soluble IL-6 and has been used in CRS, including after prior tocilizumab exposure; in the largest multicentre retrospective series to date of CAR-T-related CRS/ICANS, involving 54 patients across 6 academic centres, 75% of patients treated for CRS had improvement in CRS grade, including activity in tocilizumab-exposed cases, although the supporting evidence remains retrospective and limited to haematological settings. 67
Corticosteroids
Corticosteroids provide broad suppression of cytokine signalling and endothelial inflammation and remain a cornerstone for CRS, being recommended for grade 2 or higher CRS, or when CRS co-exists with immune effector cell-associated neurotoxicity where IL-6R blockade is not indicated. 60 Typical regimens include dexamethasone (e.g. 10 mg IV repeated according to severity) or methylprednisolone (1–2 mg/kg/day) for severe, rapidly progressive cases, followed by prompt de-escalation once clinical improvement is achieved, to minimise infectious and metabolic complications. 43 In TCEs, corticosteroids are also commonly embedded as prophylaxis (premedication during step-up dosing) and as part of treatment algorithms for CRS. While early steroid use was historically avoided due to concern about attenuating anti-tumour immunity, several cohorts suggest that short-course, early corticosteroids can reduce progression to high-grade CRS without clearly compromising clinical outcomes or CAR-T cell kinetics, supporting their judicious use when clinically required. 68
IL-1 blockade
IL-1 comprises a family of related cytokines, including agonists (such as IL-1α and IL-1β), receptor antagonists and an anti-inflammatory cytokine. 69 Among these, IL-1β is the principal CRS-relevant effector, acting as an early upstream trigger that drives myeloid activation and amplifies downstream cytokine signalling. 70 Consistent with this, preclinical CAR-T cell models have shown that monocyte/macrophage-derived IL-1 is an early driver of toxicity and amplifies a downstream IL-6-mediated cytokine release, ultimately leading to endothelial activation, capillary leak and organ dysfunction. 70 By antagonising the IL-1 receptor, the upstream signalling is inhibited, thereby reducing propagation of the downstream pro-inflammatory cytokine cascade. Anakinra, a recombinant IL-1 receptor antagonist, competitively inhibits IL-1α/β signalling and therefore attenuates fever, vascular leak and myeloid activation without directly suppressing T-cell cytotoxic function. 71 Moreover, anakinra can cross the blood–brain barrier, supporting a potential neuroprotective role. Translational studies in animal models and humans have shown that its peripheral administration determines cerebrospinal fluid concentrations within the range required to antagonise central IL-1 signalling, providing pharmacokinetic evidence for effective IL-1 blockade within the CNS.72,73 To date, no prospective studies have evaluated IL-1 blockade for CRS in patients with solid tumours, and its potential role in this setting therefore remains inferred from preclinical evidence and experience in haematological malignancies.
Consistent with this, in haematological malignancies, anakinra has been increasingly incorporated into the management of immune-effector cell toxicities, particularly in high-grade or steroid-refractory CRS and ICANS. 63 In a retrospective multicentre study of patients treated with CAR-T cells, high-dose intravenous anakinra (up to 12 mg/kg/day) for refractory CRS/ICANS was well tolerated, with a median time to CRS/ICANS resolution of 7 days from anakinra initiation. 74
Although these data derive from haematological malignancies, they provide an important mechanistic rationale for exploring prophylactic IL-1 blockade in solid tumours.
Together with Anakinra, which is a short-acting IL-1 inhibitor, canakinumab, a fully human monoclonal antibody targeting IL-1β, represents a long-acting IL-1-blocking strategy; in hyperinflammatory states, it is typically administered 4 mg/kg subcutaneously. 75 Although it is not established as standard therapy for CRS, canakinumab has been investigated in cytokine-storm syndromes in COVID-19 pneumonia, where prospective studies suggest that IL-1β blockade can reduce systemic inflammation with a manageable safety profile.76,77 These observations support further exploration of IL-1 blockade, anakinra for rapid modulation and canakinumab for sustained IL-1β suppression, as a mechanism-based strategy for CRS in solid tumours, particularly in patients at high risk of severe neurotoxicity or refractory CRS.
Janus kinase–signal transducer and activator of transcription inhibitors
The Janus kinase-signal transducer and activator of transcription (JAK–STAT) pathway is a key intracellular signalling cascade involved in immune regulation and inflammation, integrating signals from over 50 cytokines and growth factors, including IL-6, IL-2, IL-4, IFN-γ and TNF-α. 78 JAKs are covalently bound to cytokine receptors and become activated upon cytokine binding. This activation leads to phosphorylation of STAT proteins, which then translocate to the nucleus to regulate gene transcription. 78
In CRS, IL-6-driven activation of the JAK–STAT pathway represents a central mediator of inflammation, providing a strong rationale for therapeutic inhibition of this pathway. 79 Ruxolitinib, an oral JAK1/2 inhibitor, suppresses STAT1 phosphorylation and has demonstrated both tolerability and efficacy in small cohorts of patients with secondary HLH and CAR-T-related CRS, including cases refractory to corticosteroids.80–82 Beyond IL-6 blockade, JAK inhibition reduces the release of other pro-inflammatory cytokines that signal through JAK1, including TNF-α and IFN-γ.83,84
The selective JAK1 inhibitor itacitinib has also shown promise in prophylaxis of CRS. In a randomised study, patients receiving anti-CD19 CAR-T therapy and itacitinib 200 mg twice daily had significantly lower rates of grade ⩾2 CRS and ICANS compared with placebo (p = 0.003). 85 Preclinical data further suggest that JAK1 inhibition may have therapeutic potential in CRS secondary to T-cell bispecific antibodies. 86 However, in a clinical study combining ruxolitinib with the CD123 × CD3 bispecific DART flotetuzumab, no clear improvement in clinical CRS severity was observed despite marked reductions in circulating cytokine levels, raising the possibility that circulating cytokine changes alone may not be a reliable surrogate for clinical CRS severity. 87
At present, there are no published data evaluating the efficacy of JAK-pathway inhibitors for CRS associated with solid tumour therapies.
GM-CSF neutralisation
GM-CSF plays a key role in the development of CRS through myeloid cell activation, resulting in IL-6, IL-1β and TNF-α secretion. 63 GM-CSF neutralisation is therefore an attractive therapeutic strategy to dampen inflammatory responses and maintain effector T cell responses simultaneously. Lenzilumab, a GM-CSF neutralising monoclonal antibody previously demonstrated a significant reduction in CAR-T–related CRS and neuroinflammation and reduced circulating cytokines in COVID-19 pneumonia.88,89 Similarly, mavrilimumab, an anti-GM-CSF receptor α antibody, reduces myeloid cell-derived cytokine release with benefit in severe COVID-19 pneumonia and systemic hyperinflammation. 90 However, direct clinical evidence supporting GM-CSF neutralisation for CRS remains limited, as the available data for lenzilumab and mavrilimumab derive largely from COVID-19-associated hyperinflammation rather than oncology-specific CRS.
In summary, GM-CSF neutralisation offers a promising approach to treat CRS while preserving anti-tumour immunity and further research regarding its potential efficacy in this setting is warranted. Available data suggest that its inhibition is considered safe with mild reversible adverse events, although careful attention should be given to increased risk of infection.
TNF-α inhibitors
TNF-α is another early pro-inflammatory cytokine implicated in CRS pathophysiology. In a CD3-bispecific model, Li et al. showed that T cell-derived TNF-α is the key trigger for myeloid activation, with monocytes and macrophages mainly implicated in release of IL-6 and IL-1β. Accordingly, TNF-α blockade markedly reduced IL-6 and IL-1β release and attenuated the cytokine cascade, while largely preserving CD3-bispecific-mediated T-cell cytotoxic function.91,92
Two TNF-inhibitors have been developed so far. Etanercept, a soluble TNF receptor fusion protein that binds circulating TNF, and infliximab, a monoclonal antibody directed against TNF-α. Both are well established in rheumatologic and inflammatory diseases, but their use in CRS has been largely confined to CAR-T cell-associated toxicities and refractory hyperinflammatory syndromes, with solid tumour data remaining limited.92,93 Some reports and small series suggest TNF-α inhibition may be a useful adjunct in selected steroid- and tocilizumab-refractory ICI-associated CRS, typically as part of multi-agent rescue strategies.94–96
The main side effects related to TNF-α inhibitors are an increased risk of serious infections, including reactivation of latent tuberculosis and opportunistic viral or fungal infections, as well as potential exacerbation of demyelinating disorders.97,98 These risks are particularly relevant in patients with solid tumours, who are often lymphodepleted and concurrently exposed to high-dose corticosteroids or multiple immunosuppressive agents for CRS management. Thus far, published experience with TNF-α blockade for CRS has not reported any additional concerning safety signals; however, the available data are limited.
IFN-γ neutralisation
IFN-γ is a key mediator of systemic inflammation and macrophage activation and is implicated in the pathogenesis of CRS and related hyperinflammatory conditions such as MAS/HLH.99,100 Emapalumab, a humanised anti-IFN-γ monoclonal antibody, is approved by the US Food and Drug Administration for the treatment of refractory, recurrent or progressive primary HLH and has been investigated for use in other cytokine-driven disorders, including CRS. 101
In a retrospective paediatric series, emapalumab achieved marked reductions in circulating cytokine levels and clinical improvement in children with refractory CRS secondary to anti-CD19 CAR-T therapy, without evidence of impaired CAR-T cell efficacy. 100 Similar findings were reported in a subsequent series of eight adults with severe toxicities refractory to anakinra, tocilizumab, siltuximab and corticosteroids. 102 Although prospective evidence is lacking, a recent case report described successful use of emapalumab for multi-drug-refractory CRS after PSCA-targeted CAR-T cell therapy in metastatic castration-resistant prostate cancer, with rapid haemodynamic improvement and no clear signal of impaired CAR-T cell persistence, suggesting that IFN-γ blockade may be feasible in selected solid tumour settings. 103
The approved starting dose in HLH is 1 mg/kg twice weekly, however, dosing in the context of CRS remains variable, with reported regimens ranging from 1 to 10 mg/kg administered as single or multiple doses, underscoring the absence of established CRS-specific guidance.99,102,104 To date, the role of emapalumab in the management of CRS associated with solid tumours or non-immune effector cell therapies remain largely unexplored.
Challenges and knowledge gaps
Although progress has been made in understanding and managing CRS, important unmet challenges remain in patients affected by solid tumours and treated with immune-activating therapies, where data are limited.
Biomarker development
A major challenge is the lack of validated biomarkers to predict, detect and monitor CRS in patients with solid tumours. In haematological CAR-T cell therapy, higher baseline tumour burden, elevated ferritin and CRP, and early post-infusion increase in cytokines such as IL-6, IFN-γ and IL-10 correlate with CRS severity and have been used to build early prediction models.105,106 By contrast, analogous predictive models in solid tumours remain poorly defined. Retrospective series and reviews of CRS in solid tumours, predominantly in patients receiving ICIs and, to a lesser extent, emerging TCEs, postulate that a high baseline inflammatory burden, reflected by elevated CRP, ferritin, LDH or pro-inflammatory cytokines and extensive disease, particularly with liver metastases, may be associated with more severe events.44,95,107 However, these data derive from small case series have not yet been prospectively validated in clinical trials.
In the TCE setting, the most detailed biomarker data come from tebentafusp in metastatic uveal melanoma. Early-phase and phase III studies, and subsequent translational analyses, have consistently shown a characteristic cytokine release after dosing – with marked transient increases in IFN-γ, TNF-α, IL-2 and IL-6, together with interferon-induced chemokines such as CXCL9-11 – was temporally associated with CRS episodes and pharmacodynamic T-cell activation. Interferon-driven chemokine signatures have also been correlated with clinical benefit, underscoring the shared pathways underpinning toxicity and anti-tumour efficacy.61,108,109 Nevertheless, no prospectively validated cytokine thresholds or baseline predictors have yet been established to stratify CRS risk with tebentafusp or other TCEs. Furthermore, the identification and validation of predictive biomarkers would be crucial for differentiating CRS from infection, tumour-related fever or early haemophagocytic syndromes.
In solid tumours, case reports and small series of ICI-related CRS consistently describe marked elevations in IL-6, IFN-γ and ferritin at the time of CRS, often exceeding levels seen during infectious episodes in the same patient and falling rapidly after corticosteroids or IL-6 blockade.95,110 Serial ferritin and IL-6 trends therefore represent pragmatic markers for emerging CRS, whereas procalcitonin appears more specific for bacterial sepsis and may help to distinguish infection from immune-mediated hyperinflammation. 111 By contrast, CRP rises in both settings and therefore has limited specificity for distinguishing between them, consistent with the broader literature evaluating biomarkers of infection and systemic inflammation. 112 Interestingly, Daoudlarian et al. 113 recently conducted a biomarker-directed study of 35 patients with different solid tumours who developed hyper-inflammatory conditions during ICI therapy, including immune-related HLH (irHLH), immune-related CRS (irCRS) and sepsis, and used deep immune profiling to identify discriminatory signatures. Ferritin and hepatocyte growth factor (HGF) each achieved 100% positive and negative predictive values for distinguishing irCRS from irHLH, while serum CXCL9 levels identified patients likely to require escalation beyond corticosteroids. Notably, all 12 patients with grade 3 steroid-refractory irCRS achieved rapid clinical and biochemical remission within 24 h of tocilizumab, without loss of tumour control.
Emerging evidence supports a multi-layered biomarker tool for CRS in solid tumours: baseline inflammatory and disease-burden markers (IL-6, CRP, ferritin, LDH, liver metastases) for risk stratification, dynamic on-treatment changes and expanded cytokine panels (including HGF, CXCL9, sIL-2Rα, IL-10 and others) to differentiate overlapping hyperinflammatory syndromes and guide escalation to targeted therapies. However, almost all these potential biomarkers are derived from small series, case reports or ICI cohorts, and none has yet been prospectively validated as a predictive or diagnostic tool specifically in solid tumours.
Optimal timing, combinations and sequencing of cytokine blockers
When CRS occurs, a key unanswered question is how best to manage cytokine-directed therapies to control toxicity without undermining anti-tumour immunity.
In practice, management in solid tumours largely mirrors haematological CAR-T cell protocols, with a stepwise escalation from corticosteroids and IL-6 receptor blockade to, in refractory cases, IL-1 or TNF inhibition, despite the absence of solid tumour-specific trials defining the optimal sequence or thresholds for escalation.63,114
Preclinical models suggest that early multi-cytokine targeting could be more effective than single-agent therapy. In humanised mice with high leukaemic cell burdens, combined IL-1 receptor antagonism and IL-6 blockade more completely abrogated the inflammatory cascade and neurotoxicity experienced after CAR-T administration than IL-6 inhibition alone. 115 Despite this, combination cytokine blockade (e.g. tocilizumab plus anakinra or TNF inhibitors) has been used almost exclusively as rescue for refractory CRS or CRS/HLH overlap in small series and has not been systematically evaluated as upfront or pre-emptive therapy in solid tumours.74,116 Such an approach may also entail clinically relevant trade-offs, including increased immunosuppression and a higher risk of infection, while its effect on subsequent anti-tumour efficacy remains uncertain.
This uncertainty is especially relevant because the same cytokine networks that drive CRS also may represent a pharmacodynamic surrogate of T-cell activation.
Early clinical trials of tebentafusp showed that markers of T-cell activation, including IFN-γ-driven chemokine signatures and transient cytokine elevations, were associated with improved survival, underscoring that the same inflammatory pathways may reflect both treatment-related toxicity and effective anti-tumour immune activation. 108 Accordingly, an important unresolved question is whether indiscriminate suppression of early CRS, particularly with more intensive or pre-emptive cytokine-directed therapy, might blunt pharmacodynamic T-cell activation and, in turn, compromise anti-tumour efficacy.
Prophylactic and risk-adapted strategies in solid tumours
A recent real-world study on patients (n = 119) with multiple myeloma, demonstrated that a single dose of tocilizumab before the first dose of bispecific antibodies reduced the incidence and severity of CRS without negatively impacting the efficacy of the therapeutic agent. 117
However, the generalisability of these findings to solid tumours remains uncertain, as CRS biology may differ substantially across disease settings owing to differences in tumour microenvironment, antigen distribution and the kinetics of immune-cell engagement.
In solid tumours, prophylactic cytokine blockade with IL-6 or IL-1 inhibitors remains largely unexplored.
In addition to cytokine-directed drugs, dosing and administration strategies are central to CRS prevention. Preclinical models suggest that step-up dosing and, in some bispecific agents, subcutaneous administration can attenuate peak cytokine levels and reduce CRS risk while maintaining pharmacodynamic T-cell redirection.118,119 To date, prophylactic strategies have focused predominantly on premedication with corticosteroids rather than preventive biologic cytokine blockade. With the DLL3 × CD3 TCE tarlatamab in extensive-stage SCLC, a step-up dosing schedule combined with corticosteroid and antipyretic premedication has been associated with a shift in CRS towards predominantly low-grade events occurring early in treatment, and this strategy has been incorporated into the approved regimen. 6 Similarly, for tebentafusp in metastatic uveal melanoma, CRS mitigation relies on a weekly step-up schedule, in-patient monitoring for early infusions and supportive care; although IL-6 and other cytokines rise sharply after dosing, pre-emptive IL-6 or IL-1 blockade has not been routinely employed in trials or current practice.5,61
Risk stratification and personalised prophylactic strategies are likely to become increasingly important components of CRS management in solid tumours. In principle, clinical and biomarker-based risk calculators integrating tumour burden, baseline inflammatory markers and performance status could identify patients who might benefit from pre-emptive interventions, while allowing lower-risk patients to be managed with standard step-up regimens alone. However, such models have not yet been validated for CRS in solid tumours, and there are no data to support routine prophylactic cytokine blockade before T-cell-redirecting therapies initiation.
Next-generation TCEs
TCEs are reshaping the treatment paradigm in solid tumours, but next-generation designs are needed to maintain efficacy while improving tolerability and thereby extending their use to a broader patient population. Structural engineering strategies to optimise TCE architecture and mitigate immune-related toxicities are now beginning to emerge.
An intriguing strategy is the development of TCEs incorporating low-affinity anti-CD3 binding arms. TNB-585, a PSMA × CD3 bispecific TCE tested for metastatic castration-resistant prostate cancer, combines a low-affinity anti-CD3 arm with anti-PSMA binding and has shown in vitro and ex vivo that it can retain potent tumour cell killing while inducing lower cytokine release compared with a higher-affinity anti-CD3 PSMA bispecific antibodies. 120
Another strategy to improve the therapeutic index of TCEs is the development of conditionally activated, ‘masked’ compounds that are selectively unmasked in the tumour microenvironment. 121 Precision-activated TCEs, often referred to as XPAT proteins, use protease-cleavable masking fragments to sterically block CD3 and tumour-associated antigen binding in the circulation, thereby limiting on-target/off-tumour toxicity and systemic cytokine release. 121 Upon exposure to tumour-associated proteases, these masking fragments are cleaved, allowing the CD3 × tumour-antigen engager to become fully active within the tumour microenvironment. In preclinical studies, precision-activated TCEs have demonstrated a prolonged half-life, enhanced tumour selectivity and reduced systemic cytokine release compared with unmasked TCEs. 121 VIR-5818, formerly AMX-818, is a HER2xCD3 XPAT TCE that has entered early clinical evaluation and is currently being tested as monotherapy and in combination with pembrolizumab in patients with HER2-positive tumours across multiple solid tumour types (ClinicalTrials.gov identifier: NCT05356741).
Similarly, next-generation TCEs have been developed to deliver tumour-specific T-cell cytotoxicity while simultaneously modulating immune checkpoints, through PD-1/PD-L1 inhibition. 122 In this design, protease-cleavable PD-1/PD-L1 heterodimer is used as a steric mask to inhibit the CD3 binding in peripheral tissues, thereby limiting off-tumour T-cell activation and systemic toxicity.
By contrast, within the tumour microenvironment, tumour-associated proteases cleave the linker to unmask CD3 binding, converting the molecule into an active tri-specific compound that combines T-cell redirection, PD-1/PD-L1 axis modulation and tumour-antigen targeting.
This innovative engineered platform has shown broad applicability across different tumour-associated antigens, including HER2, underscoring its potential as a versatile platform for next-generation–activated TCEs.122,123
Standardisation of CRS definitions and grading in clinical trials
In solid tumours, as described, CRS induced by T-cell-redirecting therapies is reported mostly in small, heterogeneous early-phase trials, with substantial variability in grading and attribution. 20 Prospective, multicentric clinical trials will enable systematic characterisation of CRS incidence, grading, management and provide solid evidence for validating biomarker-based risk scores and risk-adapted prophylactic strategies. 124
Similarly, an important unmet need is the adoption of standardised CRS grading in clinical trials. The ASTCT consensus criteria, originally developed in the CAR-T setting, were specifically designed to harmonise CRS and neurotoxicity grading across immune-effector therapies, yet ICI and TCE studies in solid tumours often use CTCAE terminology or trial-specific definitions to report CRS and infusion-related events, rather than uniformly applying ASTCT grading.14,124 This heterogeneity complicates cross-trial comparisons and obscures the true burden of CRS in solid tumours, a limitation that will become even more critical as T-cell-redirecting therapies, and particularly TCEs, move towards routine clinical practice.
Prospective implementation of harmonised CRS grading will be essential to accurately characterise toxicity across trials, enable meaningful cross-study comparisons and inform the design of future studies testing risk-adapted prophylaxis and rational cytokine-blocking combinations.
Conclusion
CRS has emerged as a clinically relevant, yet insufficiently characterised, toxicity of T-cell-redirecting therapies in solid tumours. Mechanistic insights from preclinical models and early clinical trials support a mechanism-based approach to CRS management, using IL-6, IL-1 and TNF-α blockade, alone or in combination with corticosteroids, to attenuate hyperinflammation while preserving anti-tumour T-cell function. Next-generation TCE designs, that incorporate built-in attenuation or conditional activation, could potentially offer additional opportunities to widen the therapeutic window while reducing the toxicity burden. A major limitation of the current literature is that most available evidence is either extrapolated from haematological malignancies or derived from small, heterogeneous series in solid tumours.
Priority areas for future research include biomarker-driven risk stratification, optimisation of timing and sequencing of cytokine inhibitors and the prospective evaluation of prophylactic strategies in high-risk patients.
Establishing standardised CRS grading and dedicated registries in solid tumour trials will be crucial to accurately quantify risk, refine management algorithms and ultimately enable broader, safer use of T-cell-redirecting therapies in solid tumours.
Supplemental Material
sj-docx-1-tam-10.1177_17588359261457552 – Supplemental material for Cytokine release syndrome in solid tumours: mechanism-based therapeutic strategies for prevention and management
Supplemental material, sj-docx-1-tam-10.1177_17588359261457552 for Cytokine release syndrome in solid tumours: mechanism-based therapeutic strategies for prevention and management by Federico Monaca, Rachel Woodford, Kroopa Joshi, Fiona Thistlethwaite, Kok Haw Jonathan Lim and Igor Gomez-Randulfe in Therapeutic Advances in Medical Oncology
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.