
Abstract
Introduction
This feasibility study aimed to (i) develop a clinical protocol using a long-term potentiation-like repetitive stimulation protocol for transcutaneous electrical nerve stimulation in patients with upper limb complex regional pain syndrome and (ii) develop a research protocol for a single-blind randomised controlled trial investigating the efficacy of transcutaneous electrical nerve stimulation for complex regional pain syndrome.
Methods
This small-scale single-blind feasibility randomised-controlled trial planned to randomise 30 patients with upper limb complex regional pain syndrome to either a variant of transcutaneous electrical nerve stimulation or placebo transcutaneous electrical nerve stimulation for three weeks. Stimulation comprised 20 pulses over 1 s with a non-stimulation interval of 5 s, a so-called repetitive electrical stimulation protocol following the timing of long-term potentiation. Pain, function and body image were measured at baseline, post-treatment and at three months follow-up. At three months, participants were invited to one-to-one interviews, which were analysed thematically.
Results
A transcutaneous electrical nerve stimulation protocol with electrodes applied proximal to the area of allodynia in the region of the upper arm was developed. Participant concordance with the protocol was high. Recruitment was below target (transcutaneous electrical nerve stimulation (n = 6), placebo (n = 2)). Mean (SD) pain intensity for the transcutaneous electrical nerve stimulation group on a 0 to 10 scale was 7.2 (2.4), 6.6 (2.8) and 7.8 (1.9), at baseline, post-treatment and at three-month follow-up, respectively. Qualitative data suggested that some patients found transcutaneous electrical nerve stimulation beneficial, easy to use and were still using it at three months.
Conclusion
Patients tolerated transcutaneous electrical nerve stimulation well, and important methodological information to facilitate the design of a large-scale trial was obtained (ISRCTN48768534).
Keywords
Introduction
Transcutaneous electrical nerve stimulation (TENS) is commonly used for pain relief, has few side-effects and can be self-administered using relatively inexpensive devices. 1 Recent guidelines 2 identify TENS as a potential therapy for patients with complex regional pain syndrome (CRPS). However, no randomised controlled trials (RCTs) have investigated the effectiveness of TENS for CRPS.
The primary mechanism by which TENS purports to work is via the pain gate. In brief, TENS activates large diameter, low threshold mechanosensitive afferents resulting in synaptic inhibition of nociceptive transmission in the spinal cord and brainstem. 3 We propose that TENS may have additional mechanisms of action for painful conditions that involve perceptual distortions of the affected area.
Persistent pain can disrupt the representation of the painful body part in the brain of CRPS patients, often referred to as cortical reorganisation/disruption. This cortical reorganisation is hypothesised to play a role in maintaining the pain.4,5 Sensory retraining can be defined as any intervention aimed at restoring the normal body image of that area. Evidence from patients with phantom limb pain and CRPS suggest that sensory retraining of the painful body part, by physical stimulation of that body part, can normalise the disrupted body image and reduce pain.6–8 TENS, delivered in a synchronised pulsed fashion across two or more receptive fields, commonly referred to as repetitive electrical stimulation, has been used as a method of sensory retraining in individuals with diminished sensorimotor performance, such as older adults and stroke patients.9–12 Additionally, a recent non-randomised study using high-frequency repetitive sensory long-term potentiation (LTP)-like stimulation suggested it could reduce sensory impairment in patients with CRPS. 13 To date, no RCTs have investigated the effect of TENS on sensorimotor performance in patients with CRPS. However, it has been shown that TENS relieves phantom pain and facilitates perceptual embodiment of prosthetic limbs, 14 when TENS sensation is projected into the phantom limb itself. 15 We have hypothesised that TENS may be mimicking normal neural input to restore disrupted body image. 16
Study objectives.

TENS: transcutaneous electrical nerve stimulation.
Method
Study overview
This mixed-methods single-blind placebo-controlled feasibility study randomised participants with CRPS into an intervention or a placebo group.
Participants, recruitment, screening and randomisation
The study aimed to recruit 30 participants from a regional NHS hospital in the north east of England. This study was approved by the NRES Committee North East – York NHS Ethics Committee (REC approval number: 13/NE/0286). Participants were eligible for inclusion, if they were ≥18 years of age and had type-1 CRPS of the upper limb defined by the Budapest criteria 19 for ≥6 months’ duration. Those who once fulfilled the criteria, no longer did, but had ongoing pain were classed as CRPS-NOS (not otherwise specified) and were also included. Participants were excluded if they found TENS as applied in the first session unacceptable/intolerable, lacked the capacity to give informed consent, had any neurological condition (e.g. stroke) or were unable to speak English. Eligible participants provided informed consent and were randomly allocated to TENS or placebo TENS using a random number generator and pre-filled concealed envelopes by a team member uninvolved in participant contact. As this was a feasibility study an a priori sample size calculation was not undertaken, though the sample size was in keeping with recommendations that such trials should aim for approximately n = 12 in each group. 20 This study aimed to recruit 30 participants which would allow for a 20% dropout rate whilst still achieving n = 12 per group.
Preliminary rehearsal of process
Based on our previous clinical experience and direct patient involvement in the study design, it was felt that CRPS patients were unlikely to tolerate applying TENS electrodes to the painful site. Thus, we proposed TENS be applied to the upper arm, proximal to the affected site and the TENS sensation projected distally into the affected area. We easily achieved this distal projection in unimpaired participants (unpublished). This simple adaption of conventional TENS technique (e.g. placing the electrodes over the painful area) overcame this unique CRPS barrier.
Intervention
TENS, in this study, was delivered using a commercially available two-channel TENS unit (Elpha II 3000 muscle and pain stimulator, Danmeter). A detailed demonstration on self-application and a simple instruction sheet to facilitate home use was provided. Pulsed, synchronised dual channel TENS was self-administered (for 90 min) at home, daily, over a period of three weeks. The stimulation pattern described by Freyer et al. 12 was used: 20 pulses delivered over a 1-s period (20 Hz stimulation frequency) with a non-stimulation interval of 5 s. This form of stimulation is commonly referred to as repetitive electric stimulation and its primary proposed mechanism of action is cortical reorganisation rather than via the classic gate control mechanism associated with conventional TENS stimulation. A self-report diary monitored home programme concordance. All participants received usual care provided by the same physiotherapist to maximise standardisation. Usual care physiotherapy included but was not limited to advice, education, exercise, cognitive behavioural therapy, motor imagery, hand laterality recognition training, desensitizing and hydrotherapy. It excluded TENS or other electrotherapy modalities. The number of usual care physiotherapy sessions was recorded for all participants.
Placebo
The placebo group received TENS in an identical manner to the active intervention except that no electrical current was delivered, although the power light flashed pulsatingly indicating that the device was switched on. It was impossible to blind participants to receiving a strong non-painful TENS sensation. However, we attempted to markedly reduce this bias by introducing uncertainty as to whether a strong sensation was a pre-requisite for pain relief. Hence, information orientating participants to study aims and procedures were designed to raise uncertainty in the mind of the participant about whether they received an active intervention or not. This approach has been used successfully in previous studies. 21 Blinding the therapist was not possible. Similar to the participants in the intervention group, all participants in the placebo group received usual care provided by the same physiotherapist and the number of usual care physiotherapy sessions was recorded for all participants.
Clinical outcomes
Perceptual outcomes
Hand laterality recognition task
Correctly identifying the laterality (i.e. ‘left’ or ‘right’) of an image depicting a hand requires the participant to engage in the mental rotation (motor imagery) of their own limbs, 28 with optimal performance relying on an intact body schema. Previous studies have demonstrated impairment on the task in patients with CRPS.29–32 Participants were presented with images of hands on a computer screen of varying laterality, view, orientation and rotation and asked to respond as quickly and as accurately as possible. Presentation of stimuli (images) and resulting data (accuracy and response time) were controlled and logged using customised software (E-Prime, www.pstnet.com).
Bath CRPS body perception disturbances
The Body Perception Disturbance questionnaire assesses a patients’ perception of their affected limb. 33 Participants rate different aspects of perception such as feelings of ownership and attention. Five of the items are on a 0–10 scale, 2 of the items are yes/no answers and the final item is graded on a 0–1–2 scale. A total score is scaled from 0 to 57. Higher scores denote poorer perception of the limb. There is no literature identifying what is a meaningful clinical improvement on the Body Perception Disturbance questionnaire.
Research outcomes
To investigate the appropriateness of the single-blinding procedures, participants were asked to make a judgement as to which group they were randomised. The blinding assessment method asked the participants two questions. Firstly, do you believe your TENS unit was functioning properly? The five possible responses to were: (1) I am certain my TENS unit was working properly; (2) I think my TENS unit was probably working properly; (3) I have absolutely no idea whether the TENS unit was working properly or not; (4) I think my TENS unit was probably not working properly; (5) I am certain my TENS unit was not working properly. Secondly, If above you answered (3) I have absolutely no idea whether the TENS unit was working properly or not – what would you guess (1) functioning properly (2) not functioning properly. This assessment method has been previously used by our group and others in the TENS field. 35
Qualitative interviews
Interview topic guide.

Data analysis
Participant characteristics are presented as mean and one standard deviation (SD). As this was a feasibility study, the change in outcomes was investigated using descriptive statistics. The qualitative data were analysed thematically.
Results
Recruitment/eligibility
Thirteen individuals initially showed interest in participating of whom eight consented and were randomised to the TENS (n = 6) or placebo TENS group (n = 2) (Figure 1). There was a 50% dropout rate with only four participants providing data at the three-month follow-up point. The participant characteristics are shown in Table 3. The number of usual care physiotherapy sessions received by each participant ranged from 1 to 20 (Table 3).
Participant flow diagram. Participant characteristics. TENS: transcutaneous electrical nerve stimulation; PT: physical therapy.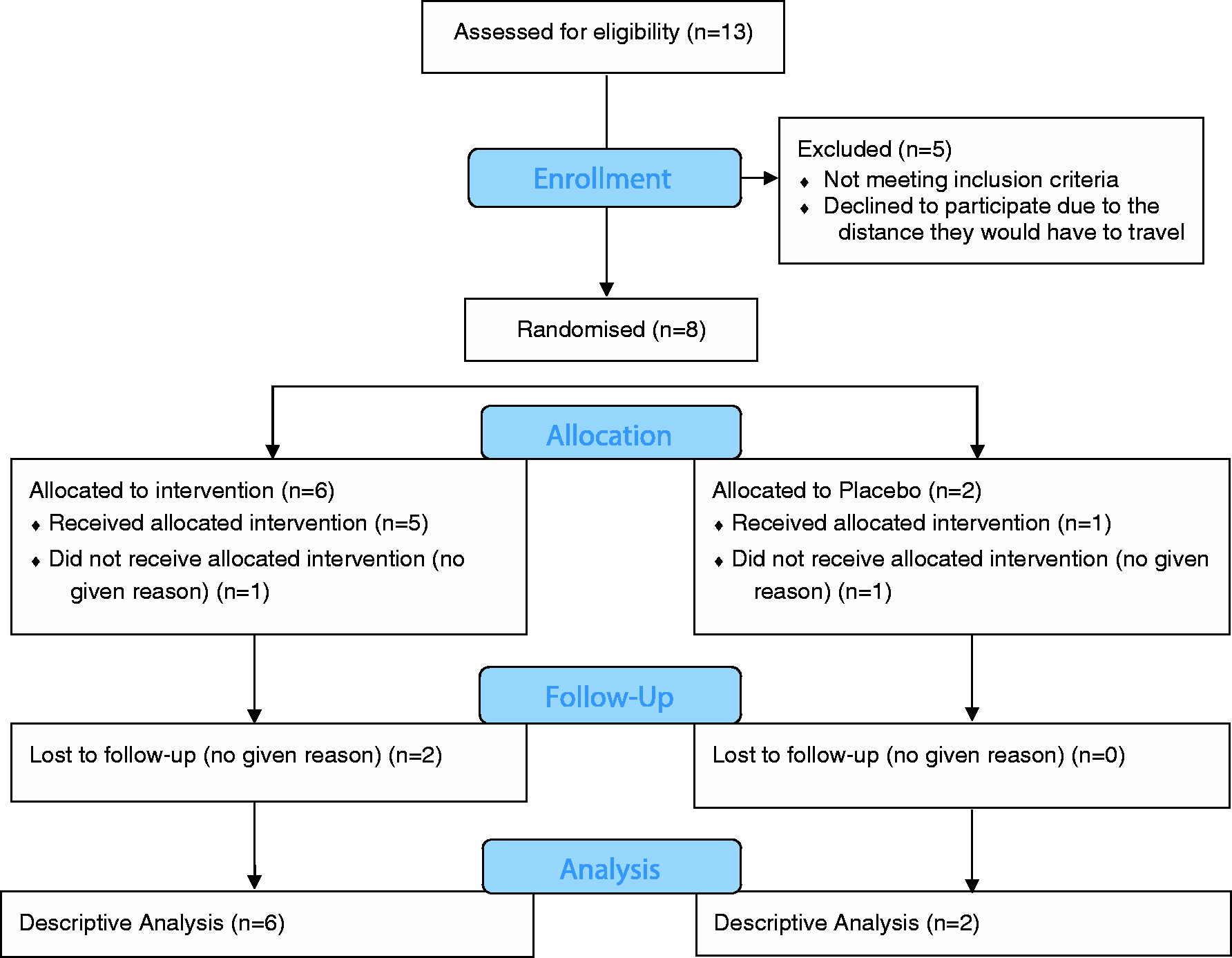

Clinical outcomes
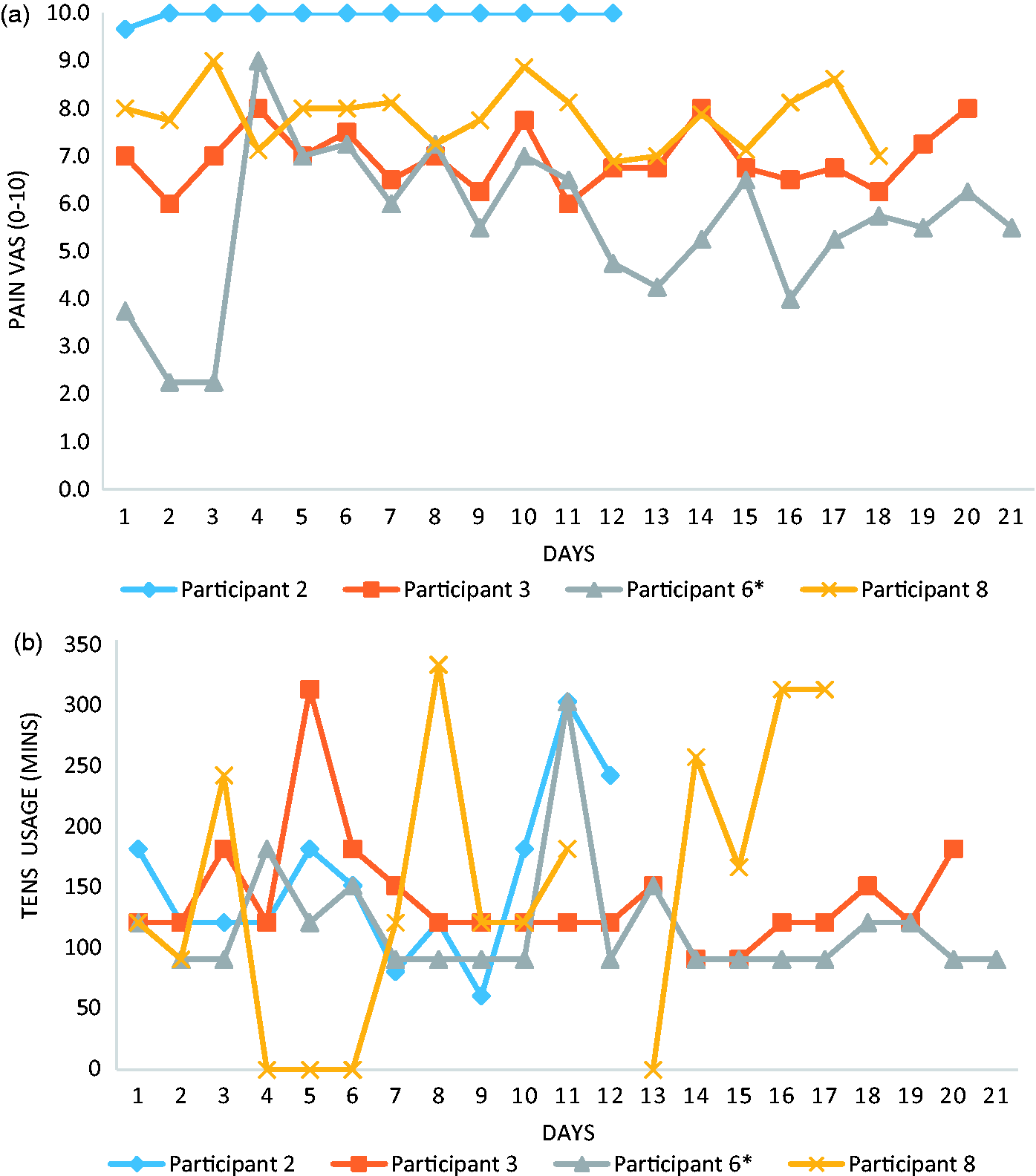
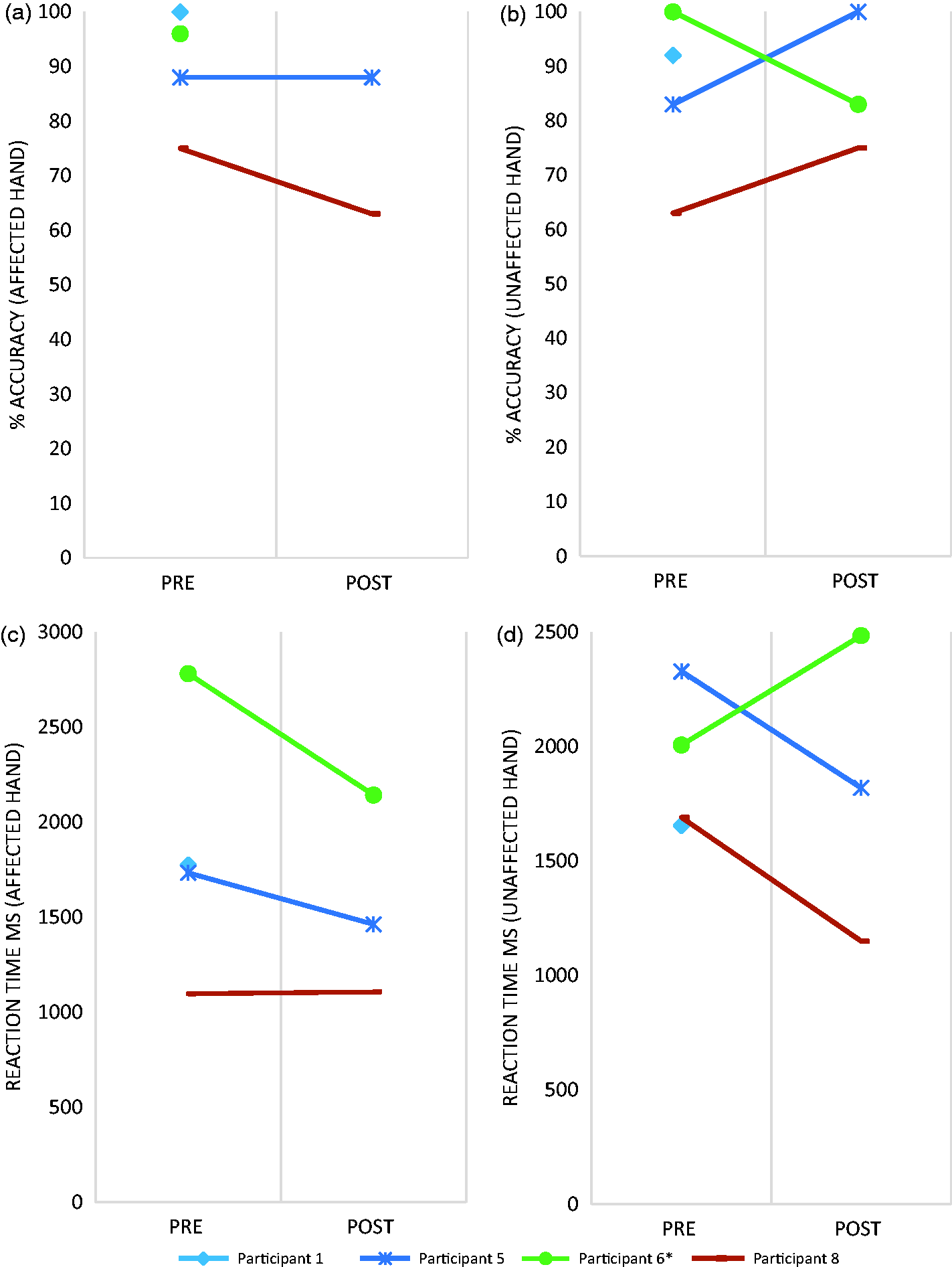
(a–c). Data are presented for each individual participant.

Perceptual outcomes
(a) Average daily pain reported by each participant who completed a diary. (b) Total TENS usage time each day for each participant who completed a diary. Missing data was left blank. (a–d). Data are presented for each individual participant.
Patient expectations and blinding
At baseline both the TENS and the placebo groups had similar levels of expectations regarding how helpful they believed TENS would be for their pain (5.8 (2.7) vs. 6.5 (0.0)), how helpful they believed sub-threshold TENS would be for their pain (5.2 (2.9) vs. 5.0 (2.1)) and how easy they thought TENS would be to use (6.4 (3.5) vs. 5.8 (5.4)).
At the three-month follow-up, all three of the participants from the TENS group who completed these questions were certain that their TENS unit was working correctly, while the one participant from the placebo group thought that the TENS unit was working correctly but was not certain.
Qualitative interviews
Four (n = 3 TENS and n = 1 placebo) participants underwent exit interviews. Two main themes were identified: (1) the level of perceived benefit and (2) the ease of use.
Of the three TENS participants, one clearly stated that they did not find TENS useful:
‘[TENS] didn’t work’ [002]
The other two TENS participants found TENS beneficial. Participant 003 stated that when the TENS was being applied it helped to take her mind off her pain and reported relief of about 50% when TENS was on but no residual benefit once removed:
‘It took my mind away from the pain while it was on…I would say about 50% [reduction in pain]’ [003]
Both of these participants were still regularly using TENS (three or four times a day (004)) but were now using it on continuous mode rather than the pulsed mode that was used in the study:
‘I did [get my own TENS machine] yeah and I keep that on the full one [continuous mode]’ [003]
There was no obvious reason identified as to why some participants found TENS beneficial and some did not. However, the participant reporting no benefit described feeling the TENS sensation in their arm/elbow, while their main painful area was their thumb. In contrast, one of the participants who found a benefit specifically reported feeling the TENS sensation in the same area as their pain.
The single participant in the placebo group who undertook an exit interview also felt that TENS had been beneficial though, they qualified that the benefit was small and felt that the effect was due to distraction.
‘I think it did decrease pain but only minimally, and I think that was possibly more due to distraction rather than actually the TENS’ [006]
This participant noted a short-term adverse event. The participant self-managed this adverse event. They did not formally report it as an adverse event within the study itself.
‘A slight increase in swelling in my hand…could be combatted with the compression glove’ [006]
All four participants reported that TENS was easy to use.
‘very easy to use’ [002]
While the application of the electrode pads and their associated wires did pose challenges, patients learned to problem solve the issues independently or with the help of a partner. I struggled at first because the wires getting in and out, obviously I can’t pull the wires totally out because it’s my right hand…but I have worked it out now I pull the pads off…or I just leave them plugged in and my partner takes them out [004]
‘Other than the fact that I can’t put the pads on my shoulder myself, it’s completely usable’ [006]
Discussion
We developed a clinical TENS protocol, where the electrode pads were applied to the upper arm, proximal to the affected site and the TENS sensation projected distally down the arm into the affected area. This simple adaption of conventional TENS technique (e.g. placing the electrodes over the painful area) was acceptable to participants who reported during interview that the device was easy to use and had no adverse effects apart from a short-term mild increase in hand swelling in one participant in the placebo group. Participant concordance to the treatment regime was high, implying that the application technique was acceptable. In addition, this feasibility study identified key methodological issues, such as pain medication usage recording, which should be taken into consideration for a future RCT.
Recruitment rates were poor. This does not appear to have been the result of inappropriate recruitment procedures per se or overly exclusive eligibility criteria. When planning this study, it was estimated that the pain department hosting the study usually had three new patients with CRPS per month, with two new patients per week attending local peripheral clinics. Thus, it was expected that recruitment goals were achievable. A number of strategies were put in place to increase recruitment including regular updates to staff to raise awareness of the study; however, it is unclear what if any effect such strategies had. During the recruitment phase, other clinical studies/clinical pathways were operating within the hospital, which may have reduced the number of individuals directed towards this study. A future large scale RCT should be multicentred and consider using existing databases such as the UK CRPS database. 37
The pain and TENS usage diary were useful outcome measures, though only four of the eight participants completed them. The reason for this poor completion is unknown. Those participants who completed diaries did so comprehensively with little missing data. The TENS usage data indicated a high level of treatment concordance with usage above the recommended 90 min in 64 of the 69 reported usage days (93% adherence). Assuming zero usage on the days that no information was inputted by the four participants, this would still indicate 76% concordance (64/84). This high usage occurred alongside relatively unchanging pain levels. Pain medication recording was inconsistent, and ultimately, the information provided was difficult to interpret and thus unusable. Other methods of monitoring pain medication usage, such as automated tracking 38 should be explored. In addition, monitoring the number of usual care physiotherapy sessions was an important step with respect to monitoring co-interventions. The number of sessions varied quite considerably between participants which may need to be adjusted for in a future fully powered trial.
There were significant challenges with both of the perceptual outcomes: the Hand Laterality Recognition Task 28 and Body Perception Disturbance questionnaire. 33 Due to technical difficulties, much of the Hand Laterality Recognition Task data was not collected. The mean (SD) Body Perception Disturbance score for all participants was 28 (15), which is higher than previously reported values for CRPS patients (21 (11)). 39 The Body Perception Disturbance scores showed no obvious pattern with scores remaining largely unchanged, and the absence of any existing data regarding minimally important difference makes it difficult to comment upon the importance of the small changes seen. The Body Perception Disturbance questionnaire contains a component, where the patient describes their mental image of their affected limb and this is drawn by the assessor/healthcare professional. This drawing influences the scoring of the questionnaire. The clinician reported that the drawing was heavily influenced by their artistic ability rather than the patient’s description, questioning the face validity of the tool. A complete assessment of the psychometric properties of the Body Perception Disturbance has not been undertaken as yet, 39 but future work may want to consider the utility of the drawing component.
The level of expectation in participants regarding both above threshold and sub threshold TENS was similar between groups suggesting our approach to make the no current placebo TENS, a viable intervention in the eyes of the participants was successful. However, regarding blinding at the three-month follow-up, there appeared to be an element of doubt about the function of the device in the placebo group that was not apparent in the TENS group implying that the blinding of participants may not have been completely successful, though it does indicate a degree of success. However, it is difficult to determine from the limited amount of data.
Within the qualitative interviews, one participant stated that TENS was of no benefit to them, this could have been related to the difficulty in projecting the TENS sensation over the painful thumb area for this patient. The other two participants who provided interviews in the TENS group both reported finding TENS beneficial. One reported a clinically relevant 50% reduction in pain when using TENS and both were still using TENS regularly at the three-month point. Interestingly, they choose to use the continuous mode of delivery compared to the burst mode used for repetitive electrical stimulation as used in this study. Furthermore, one of the participants emphasised that TENS was beneficial during stimulation, but the effects were short-lived once the TENS device had been switched off. It has been argued that TENS pain relief should only be expected when in situ and thus outcome measurement to test its effectiveness should occur at this point. 40
Study implications
The implication of this study is that a definitive trial does not, as yet, appear feasible. Recruitment is a major issue and would need to be addressed in further feasibility work before proceeding to a full trial. In addition, data collection processes need to be further enhanced, with particular emphasis on the pain medication usage diary, the body perception questionnaire 33 and the Hand Laterality Recognition Task. 28 One objective of this work was to calculate an effect size to inform a fully powered future trial. For the primary outcome measure of pain, from pre- to post-treatment, an effect size of 0.25 was shown. Using this effect size, an alpha level of 0.05, and 80% power, a sample size of 506 participants (253 per group) would be required for a fully powered trial. Given the difficulties with recruitment shown in this study, such a sample size would likely be very difficult to attain.
Study limitations
This study was limited by a number of factors already discussed above, not least the low recruitment rates and the relatively high dropout rates (50%). We did not obtain reasons for dropout, which should be explored in future work. The imbalance in the randomisation was also an issue. The randomisation had been undertaken to ensure an equal number of 15 participants would be randomised to each group; however, by chance, the random sequence initially was loaded towards assigning more to the intervention group rather than the placebo group. Blocked randomisation would have prevented this distortion and should be used in any future small-scale trials. In addition, there was limited investigation of fidelity within the study, beyond monitoring the concordance to the recommended usage dose (90 min daily) via a diary. Further checks of the quality of delivery of the intervention in the participant’s home environment would have been useful.
Conclusion
We developed a clinical TENS protocol, where the electrode pads were applied to the upper arm, proximal to the affected site and the TENS sensation projected distally down the arm into the affected area. Participants tolerated TENS well. A number of methodological issues were identified, such as low recruitment rates and inadequate data collection processes particularly regarding pain medication usage and body perception assessment. Thus, further feasibility work across multiple sites, focussing upon these methodological issues is warranted prior to undertaking a full-scale RCT.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a research grant from the British Association of Hand Therapists.
Informed consent
Written informed consent was obtained from all participants.
Ethical approval
This study was approved by the National Research Ethics Service Committee North East - York NHS Ethics Committee (REC approval number: 13/NE/0286).
Guarantor
CGR.
Trial registration
This trial was registered on the ISRCTN Registry (ISRCTN48768534).
Contributorship
CGR and DJM conceived the study. All authors were involved in protocol development. CGR, RK, and VR were involved in gaining ethical approval. RK and VR were involved in patient recruitment and data collection. CGR and MIJ analysed the data, with interpretation from DJM, HRD, CG, and TDP. CGR wrote the first draft of the manuscript. All authors reviewed and edited the manuscript and approved the final version of the manuscript.