
Abstract
Background
This study aimed to identify DNA repair–related genes causally affecting OCC risk by integrating multi-omics data using summary data–based Mendelian randomization (SMR).
Methods
SMR analyses were performed using GWAS, mQTL, eQTL, and pQTL data. Causal associations were assessed via SMR and HEIDI tests, and colocalization analysis identified shared genetic variants. Findings were validated in the FinnGen_OCC cohort.
Results
This study identified 160 methylation sites (96 genes), 30 eQTLs, and 2 pQTLs linked to OCC. Colocalization confirmed 96 mQTLs, 20 eQTLs, and 2 pQTLs (GPN1, HNRNPAB). Five mQTLs (ANK1, HLA C, TERT, TUBB, VHL) replicated in FinnGen. Integration of mQTL and eQTL data highlighted VHL, TP73, and JMJD1C, implicating DNA methylation–mediated regulation. Multi-omics and tissue analyses identified VHL as the most consistent risk gene, supported by colocalization, TCGA HNSC transcriptomic validation (P < 0.01), and DepMap functional data. GEN1 and POU5F1 expression in salivary gland tissue also associated with OCC risk.
Conclusion
This multilayer SMR study reveals causal links between DNA repair–related genes and OCC, highlighting VHL as a key driver and suggesting targets for early detection and precision therapy.
Introduction
Oral cavity cancer (OCC) is among the most common malignant tumors worldwide and represents a substantial global health burden due to its high incidence and mortality. According to GLOBOCAN estimates, over 300,000 new OCC cases are diagnosed each year, resulting in approximately 145,000 deaths annually. 1 The major clinical features of OCC—pain, ulceration, and oral mucosal masses—severely impair patients’ speech and swallowing abilities, as well as their quality of life. 2 Although standard treatments such as surgery, radiotherapy, and chemotherapy remain the mainstay of care, they are associated with low five-year survival rates and a high risk of recurrence. 3 Therefore, clarifying the molecular basis of OCC is essential for improving early diagnosis and developing targeted therapies.
DNA repair mechanisms play a crucial role in maintaining genomic stability and preventing carcinogenesis. The failure of DNA damage repair leads to the accumulation of somatic mutations that drive malignant transformation and tumor progression. 4 Accumulating evidence demonstrates a strong association between abnormal DNA damage response (DDR) gene expression and OCC. For instance, ATM and γH2AFX expression levels are significantly higher in oral squamous cell carcinoma (OSCC) than in low-risk dysplasia or normal mucosa, and their co-expression correlates with enhanced malignant potential. 5 Multivariate analyses further indicate that high ATM and γH2AFX expression significantly increase the risk of malignant transformation. Likewise, imbalances in mismatch repair proteins, such as defective MLH1 or MSH2, can exacerbate DNA damage and influence tumor proliferation and patient prognosis in OCC.6,7
Recent research has also highlighted the regulatory roles of non-coding RNAs and microRNAs in the DNA repair network and the early development of oral potentially malignant disorders (OPMDs), which serve as precursors to OSCC. For example, PPIA-guided DNA–lncRNA triplex complexes may modulate cell growth, migration, and metastasis, 8 while emerging evidence suggests that specific microRNAs regulate DNA damage repair genes and epithelial responses in premalignant oral lesions.9,10 Furthermore, the influence of localized treatments, such as lasers, on periodontal tissues and the oral microenvironment represents an emerging area of interest that may interact with these molecular networks to modulate early oncogenic progression. 11 These recent studies underscore the complex interaction between DDR pathways and non-coding RNA–mediated networks in the early steps of malignant transformation, suggesting new avenues for molecular stratification and therapy.
Genetic susceptibility also contributes substantially to OCC risk, as family history studies highlight the heritable component of disease predisposition . 12 Nevertheless, the specific DNA repair genes that causally contribute to OCC onset and progression have not been systematically determined. Traditional observational studies are limited by confounding factors and reverse causality, making it challenging to distinguish cause from correlation.
Mendelian randomization (MR) methods offer an effective strategy to evaluate potential causal relationships between genetic variants and diseases. 13 The summary data–based Mendelian randomization (SMR) approach further integrates multi-omics quantitative trait loci (QTL) data—including genome-wide association study (GWAS), methylation QTL (mQTL), expression QTL (eQTL), and protein QTL (pQTL) data—to comprehensively examine gene-to-disease causal links and reduce biases introduced by confounders.14,15 Through its HEIDI test, the SMR framework can also help exclude horizontal pleiotropy and strengthen causal inference.
In this study, we systematically integrated GWAS, mQTL, eQTL, and pQTL datasets to explore the causal effects of DNA repair–related genes on OCC risk. To ensure data reliability, we included only public European-ancestry cohorts with well-curated summary-level statistics and excluded genes or probes with missing QTL information or poor LD structure. Colocalization analyses were conducted to confirm loci harboring shared causal variants between regulatory traits and OCC signals. This multi-omics MR design aimed to identify specific DNA repair genes, such as VHL and GEN1, that may play a causal role in the pathogenesis of OCC, thereby providing robust molecular targets for early intervention and precision therapy.
Methods
Study design
This study followed the STROBE-MR reporting guidelines
13
and employed a SMR framework to explore the causal associations between DNA repair–related genes and OCC. The analytic workflow included comprehensive genetic analyses using SMR and heterogeneity in dependent instrument (HEIDI) tests, as well as colocalization analyses to identify shared genetic determinants between regulatory QTLs and OCC risk. The overall design and analytical pipeline are illustrated in
Figure S1
Data sources
A total of 1670 protein-coding genes related to DNA repair were retrieved from the GeneCards database GeneCards database (https://www.genecards.org/) using the keyword “DNA Repair”, under the filters Category = “Protein Coding” and Relevance Score > 3.
The primary discovery dataset was the OCC GWAS Catalog dataset (ID: GCST012237), including 1223 cases and 2928 controls (7,273,214 SNPs). For replication, this study used the FinnGen R10 cohort (ID: FinnGen_R10_C3_ORALCAVITY_EXALLC) comprising 832 cases and 314,193 controls (20,191,330 SNPs).
In addition, blood expression QTL (eQTL) summary data for 31,684 individuals was obtained from eQTLGen 16 and blood methylation QTL (mQTL) summary data from meta-analyses of 2 European cohorts: the Brisbane Phylogenetics Study (n = 614) and the Lothian Birth Cohort (n = 1366). 15 Blood protein QTL (pQTL) summary data for 10,708 Europeans from Pietzner et al. 17 was used. This study also obtained minor salivary gland tissue-specific eQTL data from the GTEx v8 dataset shttps://gtexportal.org/home/), which includes 838 donors and 17,382 samples from 52 tissues and two cell lines. These tissue-specific data were used to evaluate the tissue-level causal effects of candidate genes on OCC risk. For functional validation of key genes, transcriptomic data from the TCGA-Head and Neck Squamous Cell Carcinoma (HNSC) cohort were retrieved via the TCGAplot package for R to compare gene expression between tumour and adjacent normal tissue using Wilcoxon tests. Additionally, in-silico fitness effects of GEN1 knock-out were queried from the DepMap portal (https://depmap.org/portal/) under “CRISPR (DepMap Public 25Q3 Chronos score)” restricted to head-and-neck cancer cell lines.
SMR analysis
This study used the
A multi-variant extension of SMR (multi-SNP SMR, –smr-multi) was implemented to enhance robustness. This method considers all SNPs within ± 500 kb around each probe, with P < 5.0 × 10−8 and LD r2 < 0.9 relative to the top SNP. Statistical significance was evaluated by combining P-SMR and P-SMR_multi, while the HEIDI test (P > 0.01) was used to exclude pleiotropy. Results satisfying P-SMR < 0.05, P-SMR_multi < 0.05, and P-HEIDI > 0.01 were carried forward for downstream colocalization and integration analyses. 15 All SMR-derived P-values were adjusted using the Benjamini–Hochberg false discovery rate (FDR) procedure, and FDR-corrected P-values are reported throughout the results. Because the number of available pQTLs was limited, colocalization for proteins was performed whenever possible based on all statistically significant loci passing the above thresholds.
Colocalization analysis
Colocalization analyses were performed using the R package ‘coloc’ 20 to identify shared causal variants between DNA repair–related cis-QTLs (mQTLs, eQTLs, pQTLs) and OCC GWAS signals. When GWAS and QTL signals were found to colocalize, the overlapping variants were interpreted as potentially influencing OCC risk through alterations in gene-related biological processes. Five mutually exclusive hypotheses were tested 21 : H0 indicated no trait was genetically associated with the SNP; H1 and H2 indicated only trait 1 or trait 2 was associated with the SNP; H3 indicated both traits were associated with the SNP but through different causal variables; and H4 indicated both traits were associated with the SNP and shared a single causal variable.
For the colocalization analysis of mQTL-GWAS, eQTL-GWAS, and pQTL-GWAS, the colocalization region windows were set to ±1000 kb around the probe location. To allow for the colocalization of QTLs with weaker signals, QTLs with a prior probability P12 of 5 × 10−5 were considered colocalized with GWAS signals if PP.H4 > 0.5, and for a P12 of 1 × 10−5, PP.H3 < 0.5 was the criterion for successful colocalization. 22
Statistical analyses
All statistical analyses were conducted using R (version 4.3.0). The R package “ggplot2” was used for Manhattan plots, the R package “forestplot” was used for forest plots, and the plotting code for SMR Locus Plot and SMR Effect Plot was sourced from Zhu et al. 23 For TCGA expression visualizations, the ‘TCGAplot’ R package was applied; significance levels were annotated (*P < 0.05, **P < 0.01, *P < 0.001).
Results
Integration of OCC GWAS and blood DNA repair–related mQTL data
After Benjamini–Hochberg correction, no loci reached the genome-wide significance threshold (FDR < 0.05). However, based on nominal significance (P-SMR < 0.05) and consistency across platforms, the SMR analysis identified 160 suggestive methylation sites (probes), which mapped to 96 unique genes associated with OCC risk (
Table S1
, P-SMR_multi < 0.05, P-SMR < 0.05, and P-HEIDI > 0.01). Among these, 96 methylation sites (corresponding to 65 unique genes) demonstrated strong colocalization evidence (
Figure S2

Forest plots of SMR analysis results for (A) mQTLs in the GCST012237 cohort (highlighting significant colocalized genes); (B) eQTLs in the GCST012237 cohort; and (C) pQTLs in the GCST012237 cohort.
In the validation cohort (FinnGen_ORALCAVITY_EXALLC), eight methylation sites from five genes (ANK1, HLA-C, TERT, TUBB, and VHL) were replicated (
SMR validation analysis results of mQTLs in the FinnGen_R10_C3_ORALCAVITY_EXALLC cohort.
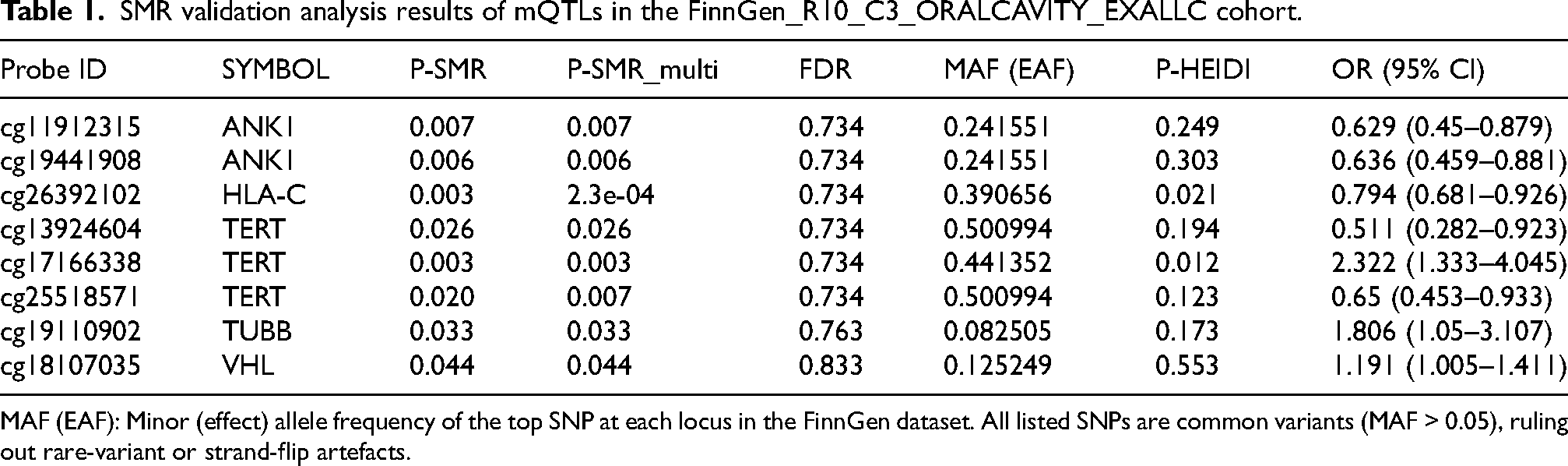
MAF (EAF): Minor (effect) allele frequency of the top SNP at each locus in the FinnGen dataset. All listed SNPs are common variants (MAF > 0.05), ruling out rare-variant or strand-flip artefacts.
Integration of OCC GWAS and blood DNA repair–related eQTL data
The SMR analysis identified 30 genes associated with OCC (
Colocalization analysis showed that the SNP colocalization windows for 20 genes contained loci associated with OCC (
Integration of OCC GWAS and blood DNA repair–related pQTL data
The SMR analysis identified 2 proteins, corresponding to genes (GPN1 and HNRNPAB), that were negatively associated with OCC (
These findings were not validated in the FinnGen_ORALCAVITY_EXALLC cohort ( Table S6 , P-SMR_multi < 0.05, P-SMR < 0.05, and P-HEIDI > 0.01).
Integration of multi-omics evidence from multiple levels
By integrating mQTL and eQTL data, the regulatory role of blood methylation was explored in the expression of key DNA repair genes in OCC. The SMR analysis revealed 4 methylation sites on 3 genes (JMJD1C, TP73, and VHL) that may play a key role in OCC (
mQTLs-eQTLs SMR analysis results.

Genomic location of the CpG site cg18107035. Functional annotation of cg18107035 showed that this CpG site is located 44 bp upstream of the VHL transcription start site (TSS200 region).

By integrating eQTL and pQTL data, the regulatory role of blood gene expression was investigated in the expression of key DNA repair–related proteins in OCC. This analysis did not yield positive results.
By integrating blood mQTL, eQTL, and pQTL data, we identified10 overlapping genes (POLR2I, CDC5L, TP73, POU5F1, INO80C, VHL, GEN1, TUBB, JMJD1C, and BMPR1A) that may have a causal association with OCC. The integrated mQTL-eQTL SMR analysis showed that TP73, VHL, and JMJD1C appeared consistently and significantly in the results ( Figure S3 ). There was no overlap between pQTL and mQTL-eQTL results. Given that colocalization analysis showed that both the VHL gene and its corresponding methylation site cg18107035 were associated with OCC and given that the association of cg18107035 with OCC risk was validated in the validation set, cg18107035-VHL was used as an example to infer the potential regulatory mechanism.
In the mQTL data, VHL corresponded to 2 methylation sites: cg18107035, which was positively associated with OCC risk, and cg16373217, which was negatively associated with OCC risk. In the eQTL data, VHL (OR = 3.285, 95% CI: 1.483–7.274) was positively associated with OCC risk. In the mQTL-eQTL data, cg18107035 was positively associated with VHL regulation (OR = 1.198, 95% CI: 1.162–1.235) (

SMR locus plots and SMR effect plots for VHL. A, B. SMR effect plot (A) and SMR locus plot (B) for VHL at cg18107035 in mQTL. C, D. SMR effect plot (C) and SMR locus plot (D) for VHL in eQTL.

Validation of VHL mRNA expression in the TCGA-HNSC cohort. Functional annotation of cg18107035 showed that this CpG site is located 44 bp upstream of the VHL transcription start site (TSS200 region).

In silico knockout analysis of GEN1 in head and neck squamous cell carcinoma (HNSC) cell lines. GEN1 displayed a Chronos gene effect score median around −0.0586 across various head and neck cancer cell lines.
Functional annotation of cg18107035 showed that this CpG site is located 44 bp upstream of the VHL transcription start site (TSS200 region;
Tissue-Specific validation
Tissue-level associations were further explored using minor salivary gland eQTL data from GTEx. GEN1 and POU5F1 remained significant (
SMR analysis of eQTLs in GTEx Minor salivary gland data with the GCST012237 cohort.

Together, these functional and tissue-specific analyses reinforce the biological relevance of VHL and GEN1 in OCC as suggested by multi-omics MR framework.
Discussion
In this study, we applied a SMR framework to investigate the potential causal roles of DNA repair genes in the development of OCC. Our integrative multi-omics analysis identified 160 mQTLs corresponding to 96 genes, 30 eQTLs, and 2 pQTLs that may be linked to OCC risk. Among these, the methylation site cg18107035 within the VHL gene demonstrated a robust positive association with OCC, supported by both colocalization analysis and independent cohort validation. Furthermore, altered expression of genes such as ANK1, HLA-C, TERT, TUBB, and GEN1 was also associated with OCC risk, highlighting their potential significance in OCC pathogenesis and progression. While no loci achieved the stringent FDR < 0.05 threshold, we prioritized candidate genes based on cross-omics consistency—specifically, overlapping mQTL and eQTL signals—and external validation using TCGA and FinnGen datasets. This strategy strengthens the biological plausibility of our findings and underscores the value of integrative approaches in elucidating the molecular pathways underlying oral carcinogenesis.
From the perspective of causal inference, analysis satisfied the main IV assumptions. Each gene's top cis-QTL (P < 5 × 10−8) served as a strong instrument, corresponding to an approximate F-statistic of 29.7 (F ≈ Z2 ≫ 10), thereby minimizing the possibility of weak-instrument bias. To further ensure robustness, we used the HEIDI test (P > 0.01) to exclude horizontal pleiotropy and to verify that each significant association was likely driven by a single shared causal variant rather than distinct linked variants. Traditional multi-variant MR approaches such as MR-Egger or weighted-median estimation were not feasible because strong linkage disequilibrium within cis regions prevents the identification of multiple independent instruments; the HEIDI test is thus the most appropriate pleiotropy control in SMR. In addition, because SMR employs germline cis-QTL genotypes that are fixed at conception, the direction of causation (molecular trait → OCC) is preserved, and the possibility of reverse causality (OCC → molecular trait) is theoretically excluded.
The VHL gene, acting as an E3 ubiquitin ligase, forms a complex with cullin-2, elongins C and B, and ring-box 1, participating in the regulation of HIF protein ubiquitination and degradation. Under hypoxic conditions, impairment of VHL gene function can lead to abnormal accumulation of HIF proteins, thereby affecting tumor development. 24 Studies have indicated that loss of the VHL gene is common in OSCC. 25 The downregulation or loss of VHL gene expression may not only promote epithelial-mesenchymal transition, enhancing the invasiveness and metastatic ability of OCC cells, but also lead to abnormal accumulation of β-catenin in the cell nucleus, activating the Wnt signaling pathway and further promoting cell proliferation and tumor formation. 26 Recent research also suggests that the VHL gene may influence the energy metabolism and growth process of OSCC by regulating the expression of glucose transporters. 27
In OSCC, there is a clear link between the VHL gene and the DDR pathway, with one study found that of 56 OSCC patients, 17 had changes in the DDR pathway and 35 had mutations in the VHL gene. 28 Loss or mutation of the VHL gene, which impairs its K63 ubiquitination function, may weaken the cellular response to DNA damage, reducing the efficiency of homologous recombination repair and DNA double-strand break repair. 29 However, this study revealed a significant positive association between the expression and methylation levels of the VHL gene and OCC risk, suggesting that high gene expression may promote the occurrence of OCC, which is inconsistent with previous study findings. Given that OCC is a heterogeneous disease, 30 patients from different regions may have different genetic backgrounds and disease subtypes. This study focused on the European population, and future validation in broader databases may be necessary. Overall, VHL gene dysfunction may lead to inactivation of the DDR pathway, thereby affecting tumor progression. These findings underscore the importance of further investigating the role of the VHL gene in OCC and provide new potential targets for its treatment.
To evaluate cg18107035 methylation in relation to VHL expression within the same tissue, we found no matched methylation–expression data for VHL in the TCGA head-and-neck cancer dataset. However, based on the Illumina HumanMethylation450 annotation, cg18107035 is located 44 bp upstream of the VHL transcription start site—within the core promoter (TSS200) region—where methylation typically exerts a strong inhibitory effect on transcription. This positional context would predict a negative correlation between cg18107035 methylation and VHL mRNA levels. Interestingly, SMR results indicate that cg18107035 hypermethylation is positively associated with OCC risk, and that increased VHL expression is also a risk factor.
This apparent inconsistency may arise from several biological and technical factors. First, the eQTL data used in our SMR analysis are primarily derived from peripheral blood, where the regulatory landscape and functional role of VHL can differ substantially from those in oral epithelial or tumor tissues. For example, VHL expression in immune cells may reflect systemic immune responses rather than tumor-intrinsic biology, a phenomenon observed for other cancer-associated genes as well. 31 Second, in advanced malignancies, compensatory or stress-induced increases in tumor suppressor gene transcripts, such as VHL, may occur without corresponding increases in functional protein, either due to translational inefficiency or the presence of non-functional or mutant transcripts. 32 Third, the Illumina methylation and expression arrays may not capture all gene isoforms equally; probe specificity can lead to detection of particular VHL isoforms that may be non-functional or differentially regulated in cancer versus normal tissues. 33 According to the Illumina HumanMethylation450 annotation, cg18107035 is located 44 bp upstream of the VHL transcription start site, within the core promoter (TSS200) region, where methylation is typically associated with strong transcriptional repression. Notably, our search of public resources such as MethSurv and cBioPortal did not identify matched methylation and expression data for cg18107035 and VHL in TCGA head and HNSC samples, precluding direct correlation analysis in tumor tissue. These results also suggest a pathogenic link between VHL-driven hypoxia signaling and oxidative stress—a hallmark of oral carcinogenesis.
Dysfunctional VHL leads to HIF-1α stabilization and metabolic reprogramming, which increase reactive oxygen species (ROS) levels. Under normal circumstances, DNA-repair mechanisms counteract ROS-induced damage; however, this study finding that both VHL and DNA-repair genes show causal associations with OCC suggests a ‘two-hit’ model, in which VHL-mediated oxidative stress combined with DNA-repair susceptibility drives malignant evolution of the oral epithelium.
In addition, within the FinnGen R10 database, we used mQTL analysis to further confirm the causal associations between the ANK1, HLA-C, TERT, and TUBB genes and OCC. Simultaneously, the GEN1 gene exhibited significant associations in both mQTL and eQTL analyses, particularly standing out in tissue-level validation. On the basis of these findings, we conducted a more in-depth investigation of these genes to clarify their causal links with OCC.
The ANK1 gene encodes ankyrin proteins, and its expression level is significantly upregulated in OSCC. 34 Moreover, ANK1 hypermethylation has been detected in the saliva of patients with oral tongue SCC. 35 The HLA-C gene, an Major Histocompatibility Complex class I gene, is primarily involved in antigen presentation and immune recognition. Studies suggest that the KIR2DL3(+)-HLA-C(+) genotype may be associated with a reduced risk of OSCC, 36 suggesting that HLA-C plays a significant role in genetic susceptibility to OCC. The TUBB gene encodes β-tubulin, a component of microtubules, and studies have shown that TUBB has a stable expression profile in OSCC cell lines and is often used as a housekeeping gene. 37 The TERT gene encodes the telomerase reverse transcriptase, responsible for telomere elongation. In OCC, especially OSCC, the frequency of TERT promoter mutations is significantly increased. 38 While the associations between these genes and OCC are gradually becoming clear, their specific impact through the DNA repair pathway requires further study.
Notably, previous candidate-gene and pathway studies in head-and-neck cancer have focused on classical DNA-repair genes such as ATM, XRCC1, and ERCC2. While many observational studies have reported associations between ATM over-expression or XRCC1 polymorphisms and malignant transformation, these findings often varied due to small cohorts and confounding environmental factors such as smoking or alcohol consumption. In contrast, the multi-omics MR framework rigorously evaluates causality and minimizes confounding, providing clear incremental value. Interestingly, SMR results prioritized VHL, TP73, and JMJD1C rather than these classical repair genes, suggesting that the molecular etiology of OCC may be driven predominantly by epigenetic dysregulation (e.g., DNA methylation) of specific repair-related targets rather than by structural variation alone. This interpretation shifts the focus from simple genetic association toward actionable regulatory mechanisms in OCC pathogenesis.
In contrast, the causal association between GEN1 and OCC is not clear. Functionally, GEN1 encodes an endonuclease that processes Holliday junctions during homologous recombination repair, directly participating in DNA repair. 39 Disruption of GEN1 may increase the accumulation of DNA damage, particularly reducing the efficiency of repair for double-strand breaks generated during the cell cycle, thereby increasing genomic instability 40 and promoting the development of OCC. Although TCGA and DepMap validations suggested that GEN1 knockout does not cause lethality but may influence cell growth, the SMR-based causal signal implies a regulatory rather than essential role, meriting further investigation at the mechanistic level.
The SMR approach employed in this study has several advantages. First, it integrated data from GWAS, mQTLs, eQTLs, and pQTLs, effectively assessing the causal associations between DNA repair genes and OCC risk. 15 Second, the SMR method can effectively exclude the influence of confounding factors, enhancing the reliability of causal inference. 41 Additionally, tissue-specific validation was conducted, further strengthening the reliability of the results. To improve interpretability, we explicitly demonstrated instrument strength (F ≫ 10) and employed HEIDI as a sensitivity test against horizontal pleiotropy. Together, these methodological refinements increase the robustness of the inferred causal links between DNA-repair genes and OCC. Moreover, recent work has proposed therapeutic targeting of DNA-repair and hypoxia-linked pathways through small RNA–based approaches. MicroRNA and siRNA therapies have been shown to modulate repair-gene expression, suppress oxidative stress, and sensitize oral cancer cells to therapy .42–44 This results highlight VHL and related DNA-repair genes as rational targets for such small-RNA-based therapeutic strategies. Collectively, these findings expand the understanding of OCC pathogenesis by linking DNA-repair dysfunction, epigenetic regulation, and oxidative stress evolution, providing new potential intervention avenues.
However, the study also has limitations. First, replication of this findings was limited: only five mQTL signals were successfully replicated in the FinnGen cohort, while no eQTL or pQTL associations reached replication significance. Second, all QTL discovery datasets were derived from individuals of European ancestry, and the FinnGen replication resource is also European; therefore, the generalizability of the results to non-European populations, such as Asian, African, or Latin-American cohorts, where OCC incidence and genetic background differ, remains uncertain and requires future population-specific validation. Moreover, the present study mainly relied on statistical inference based on summary-level omics data and lacked experimental functional validation. Future work should thus incorporate cellular and animal experiments to verify the biological mechanisms of key candidate genes and examine possible ethnic differences in their molecular regulation.
Conclusion
This study applied summary-data–based Mendelian randomization and integrative multi-omics analysis to identify potential causal associations between DNA repair genes—particularly VHL—and oral cavity cancer risk. These findings highlight DNA repair pathways as promising molecular targets for early detection and precision therapy in OCC. Further validation in diverse populations and functional studies is warranted.
Supplemental Material
sj-zip-1-cbm-10.1177_18758592261455209 - Supplemental material for Integrative multi-omics analysis-based Mendelian randomization identifies association of DNA repair–related genes with oral cancer
Supplemental material, sj-zip-1-cbm-10.1177_18758592261455209 for Integrative multi-omics analysis-based Mendelian randomization identifies association of DNA repair–related genes with oral cancer by Jingbo Wang, Weiqing Tang, Weilong Ding, Dongmin Wei, Ying Yuan and Xiaofeng Tao in Cancer Biomarkers
Footnotes
List of abbreviations
Authors’ contributions
Jingbo Wang and Weiqing Tang carried out the studies and drafted the manuscript. Weilong Ding participated in collecting data. Dongmin Wei and Ying Yuan performed the statistical analysis and participated in its design. Xiaofeng Tao participated in acquisition, analysis, or interpretation of data and draft the manuscript. All authors read and approved the final manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Shanghai Jiao Tong University Medical-Engineering Cross-Research Fund, the National Natural Science Foundation of China, Cross-disciplinary Research Fund of Shanghai Ninth People's Hospital, (grant number Grant No. YG2024QNA23, Grant No. 82502498, Grant No. JYJC202304).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
All data generated or analysed during this study are included in this published article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.