
Abstract
Two- and three-dimensional (2D and 3D) cell models derived from human stem cells have shed light on a wide range of molecular and cellular features of Huntington's disease (HD). Here we review the use of human stem cell-derived models to explore neurodevelopmental contributions to HD. We provide a timeline of key advances made in 2D and 3D model systems, ranging from differentiated monocultures to brain-like organoids and assembloids. Models along this spectrum have advanced our understanding of various disease-associated characteristics including disease protein (huntingtin) aggregation, somatic repeat instability, transcriptional dysregulation, perturbations in neurodevelopmental staging, and neural circuitry. We highlight recent findings in brain-like organoids which, despite being a relatively recent innovation, are proving to be a promising tool with which to study aberrant neurodevelopmental features of HD. All models have their limitations, and we compare and contrast the utility and limitations of various stem cell-based methods to study HD. Finally, we speculate on future advances employing advanced computational and transcriptomic methods that will expand the power of 3D model systems for the study of HD and related neurodegenerative disorders.
Introduction
Huntington's disease (HD) is primarily recognized as an adult-onset neurodegenerative disorder,1,2 yet it also involves significant neurodevelopmental alterations well before clinical symptom onset. 3 Wild-type huntingtin (wtHTT) protein regulates neuronal survival, axonal and vesicular trafficking, and brain development, with wtHTT deletion causing embryonic lethality.4–7 Given the diverse functions of wtHTT, it is not surprising that studies increasingly find that mutant huntingtin (mHTT) has adverse effects on neurodevelopment.
Comparisons between normal and HD fetal brains suggest that mHTT disrupts brain development during early stages, including the mislocalization of junctional complex proteins, which may contribute to impaired neural progenitor cell (NPC) polarity and differentiation. Specifically, fetal HD brains appear to have: (1) altered distributions of tight and adherens junction proteins; (2) reduced numbers of mitotic progenitor cells; (3) changes in the quantity and morphology of cilia at the apical surface; and (4) an increased ratio of intermediate progenitor cells to apical radial glia—two types of NPCs—in the ventricular zone of the developing cortex. 7 Research in mice, human fetal tissue, and organoids further suggests that mHTT subverts early developmental processes. The mutant protein may impair neuronal survival by disrupting the delivery of brain-derived neurotrophic factor (BDNF), decreasing cellular ATP/ADP ratios, impairing autophagy, altering ciliogenesis in striatal cells, and/or changing mitochondrial function.5–9
Neurodevelopmental perturbations in HD leave lasting footprints in the adolescent and adult brain, manifesting as structural abnormalities including reductions in intracranial volume, 10 striatal enlargement, 11 and neuronal migration defects such as periventricular nodular heterotopias—clusters of mispositioned neurons that disrupt cortical layering and architecture. 12 HD is also associated with widespread dysregulation of neurodevelopmental gene expression, particularly in cortical regions, potentially predisposing these areas to later degeneration. 3 Beyond structural changes, HD mutation carriers exhibit functional alterations such as early hyperconnectivity between the striatum and cerebellum, which diminishes over time. 11 Investigating these various neurodevelopmental features in human stem cell-derived models has emerged as a promising approach to understand the earliest molecular features of HD.
Multiple lines of evidence support the view that HD has a neurodevelopmental component, it remains unclear whether stem cell-based models consistently recapitulate these alterations and have the capacity to unveil novel disease features. This review highlights critical insights gained from studies using stem cells, stem cell-derived neural progeny, brain organoids, and assembloids to investigate the mechanisms underlying neurodevelopmental changes in HD (Figure 1). Specifically, we discuss the ability of stem cell-based model systems to reproduce disease-associated characteristics, the contributions of organoids to our understanding of HD pathogenesis, the advantages and limitations of each model system, and recent advances in computational and transcriptomic methods. Collectively, these models have yielded findings that support the view that HD is not purely a late-onset neurodegenerative disorder but also involves early neurodevelopmental disruptions that predispose the brain to later cognitive and motor impairments.
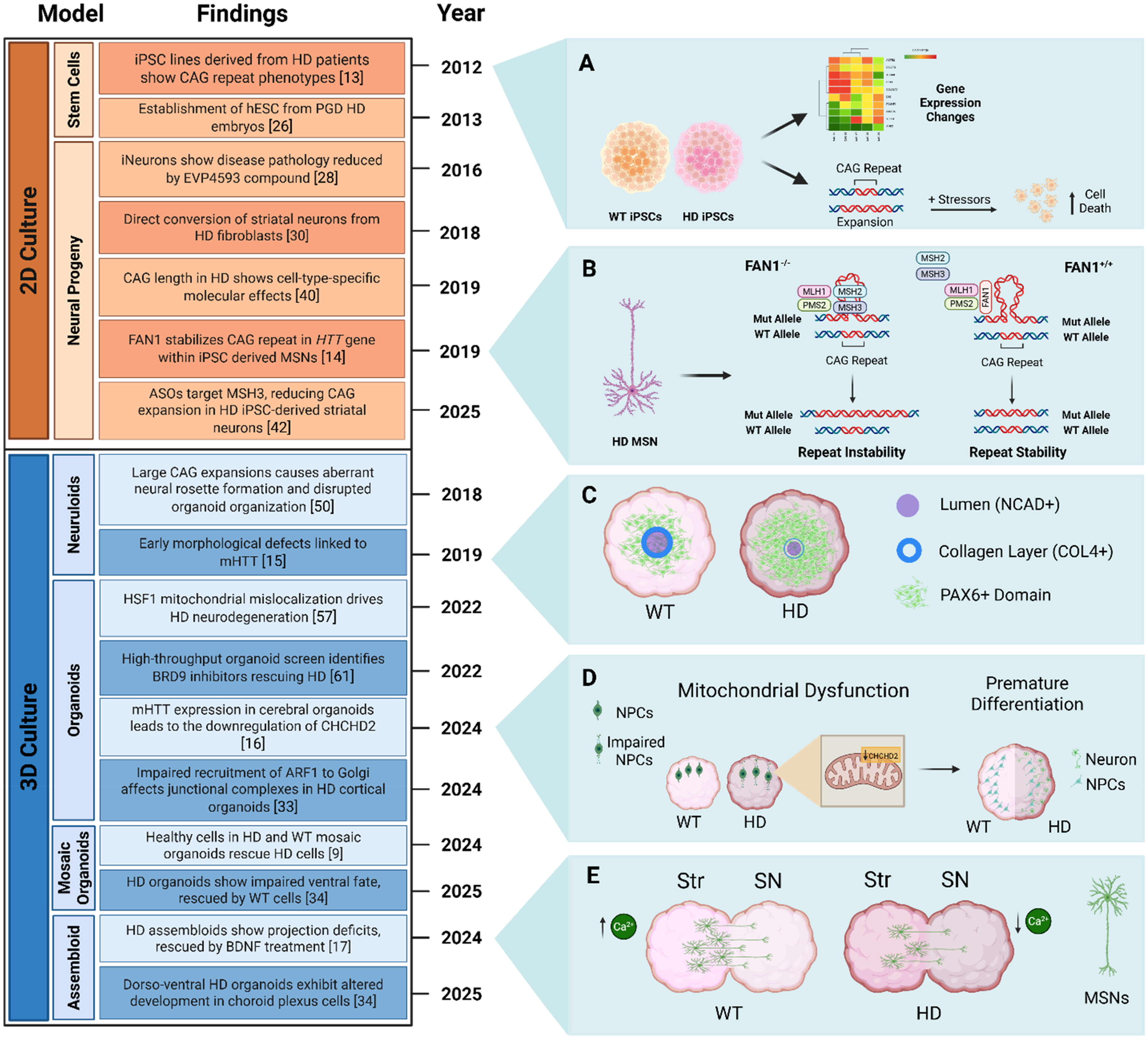
Progression of stem cell models used in HD research, demonstrating advances from two- to three-dimensional tissue culture systems and major findings across the HD field. (A) Induced pluripotent stem cell (iPSC) lines show disease-specific gene expression patterns. Lines with longer CAG repeats are more vulnerable to stress. 13 (B) Mechanism by which FAN1 stabilizes mutant HTT (mHTT) CAG repeat in medium-sized spiny neurons (MSNs): sequestering MLH1 and PMS2, preventing the binding of MSH2 and MSH3 on hairpin loops formed by larger CAG repeats, thereby stopping mismatch repair (MMR) activation and CAG expansion. 14 (C) HD neuruloids display reduced lumen size, an expanded central PAX6 domain, impaired radial symmetry, and a weakened or absent collagen layer in the central neural structure. 15 (D) Cerebral organoids with the HD-causing mutation demonstrate a downregulation of CHCHD2 (coiled-coil-helix-coiled-coil-helix domain containing 2) particularly in NPCs leading to neurometabolic failures and mitochondrial dysfunction. HD organoids also display aberrant cytoarchitecture and premature differentiation. 16 (E) HD striatum (Str) and midbrain substantia nigra (SN) assembloids display projection defects of MSNs and reduced calcium activity. 17 HD = Huntington's Disease. hESCs = Human Embryonic Stem Cells. PGD = Preimplantation Genetic Diagnosis. ASOs = Antisense Oligonucleotides. Created with BioRender.com.
Recapitulating disease-associated characteristics in stem cell models
Aberrant HTT behavior
Pathological hallmarks of HD include neurodegeneration associated with mHTT misfolding and aggregation. Although mHTT sequestration into aggregates was once considered purely toxic, it may also reflect a cellular coping response 18 and does not always correlate with pathogenicity.19,20 Whether toxic or not, aggregation has widely been used as a proxy and marker for aberrant mHTT proteostasis. Human induced pluripotent stem cells (iPSCs) and human embryonic stem cells (hESCs) carry the native human disease mutation but rarely show mHTT aggregation in their undifferentiated state,21–26 unless aged in vivo or treated with proteasome inhibitors.21,22
Similarly, iPSC-derived HD neural progenitor cells (NPCs) do not form aggregates 27 unless cultured long-term (>40 weeks) 28 or subjected to UBR5 knockdown, which impairs HTT clearance via the ubiquitin-proteasome system. 24 Neuronal differentiation allows detection of cytopathological features of HD, including the presence of nuclear indentations, increased lysosomes/autophagosomes, and oxidative stress. 28 For example, HD iPSC-derived medium-sized spiny neurons (MSNs) are more susceptible to death than control MSNs and form HTT aggregates when aged to six months or exposed to proteotoxic stress.27–29 In addition, MSNs directly reprogrammed from fibroblasts of pre-symptomatic and post-symptomatic HD patients exhibit mHTT aggregates and mitochondrial dysfunction, although MSNs from pre-symptomatic patients show reduced susceptibility to mHTT-induced toxicity. 30 A HD hESC-derived neuronal model expressing the HTT exon 1 fragment also exhibited aggregates and degeneration. 26 In another study, microRNA-196a treatment showed a neuroprotective role, leading to reduced aggregates in HD iPSC-derived neurons and reinforcing the strength of neurons in representing disease characteristics. 23 Further highlighting the disease-relevant pathology in HD MSNs, caspase-3/7 hyperactivation—enzymes known to cleave mHTT and become activated during apoptosis—was observed, but was absent in isogenic controls. 31 These findings highlight that neural differentiation enables increased detection of mHTT effects compared to the pluripotent stage.
Recently, brain-like organoids have been used to model features of HD. Brain organoids are self-organizing, pluripotent stem cell-derived three-dimensional (3D) cultures that reproduce key features of early human neurodevelopment, including brain regional patterning, neuroepithelial organization, and neuronal differentiation. 32 While they offer many advantages as a model system, organoids lack vascularization, sensory input, and the circuit integration characteristic of the intact brain. Nevertheless, organoid models provide a complex system for investigating HD pathogenesis in a human developmental context.
Most HD organoid studies have employed cerebral and cortical organoids, which correspond respectively to multiple brain regions and the developing cerebral cortex. Cerebral organoids are generated using unguided differentiation (indirect), which leverages the spontaneous differentiation and self-organization of stem cell clusters. Cortical organoids, by contrast, are produced using growth factors to direct cell fate—a process known as guided differentiation (direct). 32 Both approaches form organoids with multiple neural rosettes, each representative of a neural tube. To date, few studies employing organoids have identified aberrant mHTT behavior. According to Liu et al., cortical organoids contain soluble polyglutamine (polyQ) assemblies—non-fibrillar oligomeric complexes of mHTT—that disrupt Golgi apparatus organization and vesicular trafficking in neuroepithelial cells. 33
Organoids corresponding to different brain regions can also be combined to generate assembloids through guided differentiation.17,34 One study fused striatum (Str) and midbrain substantia nigra (SN) organoids to form Str-SN assembloids, which showed aberrant mHTT behavior including nuclear and cytoplasmic aggregates in more than 20% of MSNs. These assembloids also display neuronal loss and increased apoptosis, mirroring human HD neuropathology. 17
Alterations in protein homeostasis
The presence of disease protein aggregation in HD and other polyglutamine disorders has spawned extensive investigations of the cellular pathways maintaining protein homeostasis, also known as proteostasis. 35 The three major components of proteostasis—the ubiquitin-proteasome system, the autophagy-lysosomal system, and the molecular chaperone network—have been implicated in aspects of polyglutamine disease pathogenesis. Many investigations have shown that each of these components can counter the accumulation or aggregation of mHTT. Likewise, equally many studies employing a wide variety of model systems and human disease tissue have documented impairment in these same pathways of proteostasis.
Some of this work has been carried out in stem cells, either in their undifferentiated form or differentiated into specific cell types. Undifferentiated stem cells tend to express highly robust proteostasis machinery, safeguarding against aberrant protein behavior such as mHTT aggregation. 36 As such, it is difficult to capture aberrant protein misfolding and aggregation in undifferentiated stem cells expressing the disease protein at endogenous levels. That said, directly modulating specific components of proteostasis in stem cells can help assess which components contribute to handling a mutant protein such as mHTT. For example, manipulating proteasome activity in HD iPSCs established that the proteasome largely determines HTT levels. 24 An opportunity presented by stem cells is that scientists can compare the actions of proteostasis networks in undifferentiated stem cells versus isogenic cells differentiated into selectively vulnerable cell types, for example striatal neurons in HD. The behavior of mHTT in stems cells differentiated into MSNs suggests that the clearance of mHTT is likely achieved by processes that are more complicated than simply proteasome versus autophagy. 37
The use of brain organoids to study proteostasis networks in HD or other polyglutamine diseases is in its infancy. It holds great promise, however, for identifying how specific cell types deal with mHTT and defining the consequences of its aberrant behavior. Ubiquitin signaling pathways and molecular chaperones can vary greatly from cell type to cell type and thus may underlie some of the selective cell vulnerability in HD. Fully mature organoids maintained in culture for many months should allow scientists to query cell-specific proteostasis responses while also capturing age-dependent changes in proteostasis that explain delayed protein misbehavior in HD and related neurodegenerative diseases.
Somatic repeat instability
In vulnerable brain regions, HD individuals exhibit somatic repeat instability (SRI), a process in which the size of the HTT CAG repeat expansion progressively increases over time in somatic cells. SRI was recently recognized as a major determinant of HD progression rate and age of disease onset. 38 Expansion size has remained stable in all tested HD hESC25,39,40 and iPSC14,21,41 lines, except for an iPSC line harboring a hyperexpanded 109 repeat CAG tract. 14
Most stem cell-derived neural progeny do not exhibit SRI,39–41 with the exception of a neurosphere model—a spherical cluster of NPCs. 25 To understand the molecular drivers of SRI in HD, researchers have investigated the roles of two DNA repair factors, FAN1 and MSH3, in modulating CAG repeat expansion. HD iPSC-derived MSNs recapitulate findings from mouse models in showing that FAN1 suppresses SRI whereas FAN1 knockdown increases CAG repeat expansion. 14 Similarly, reduction of MSH3 in HD iPSC-derived striatal neurons stalled SRI, consistent with its proposed role in promoting this disease-associated characteristic. 42 The presence of SRI in organoid or assembloid models remains unexplored, representing a compelling avenue for future investigation.
Transcriptional dysregulation
Pluripotent stem cell models, including iPSCs and hESCs, reliably recapitulate some of the early transcriptional abnormalities in HD. Even in the undifferentiated state, HD iPSCs exhibit stress-induced transcriptional signatures, such as p53 pathway activation and BDNF suppression, both of which are early molecular hallmarks of disease pathogenesis. 31 The extent of transcriptional dysregulation tends to correlate with CAG repeat length, though the sensitivity of detection is influenced by assay type and culture conditions, underscoring the value of standardized protocols. 13 The occurrence of molecular abnormalities in undifferentiated cells indicates that mHTT elicits molecular effects well before the point of differentiation into neurons or other brain cell types.
Differentiated cells harboring the HD mutation, including NPCs and neurons, show hundreds of differentially expressed genes, suggesting widespread transcriptional reprogramming linked to the mutation. 40 In particular, HD NPCs demonstrate altered expression of genes involved in cell adhesion, metabolism, excitability, 13 and prolonged G1-phase duration, 43 implicating transcriptional disruption of cell cycle regulators. HD IPSC-derived three-month-old neurons show downregulation of genes associated with voltage-gated sodium currents. 44 Similar to iPSCs, fibroblast-reprogrammed HD neurons also display decreased BDNF protein expression, which correlates with increased CAG repeat length. 45
HD organoids also recapitulate transcriptional dysregulation.15,17,43 In striatal organoids, for example, DRD2 expression, which encodes the dopamine receptor D2, is selectively reduced while DRD1, encoding the dopamine receptor D1, remains unchanged, mirroring early striatal vulnerability in vivo. 17 Cerebral organoids also reproduce differentially expressed genes observed in postmortem HD brains, including downregulation of mitotic genes tied to ATM-p53 signaling—suggesting transcriptional repression of progenitor proliferation. 43
Building on these models, assembloids have further advanced our understanding of transcriptional dysregulation by capturing interactions between multiple brain regions. Specifically, Str-SN assembloids recapitulate some aspects of early stem cell findings, including partial rescue of network deficits with BDNF, highlighting endogenous trophic deficiency. 17 In addition, a HD dorso-ventral assembloid model revealed dysregulated expression of several inhibitory neuronal markers, 34 supporting the notion that transcriptional dysregulation is ubiquitous and shared across HD two-dimensional (2D) and 3D models.
Functional and developmental capabilities of 2D and 3D HD models—utility, strengths, and limitations
Multicellular environment/circuitry
iPSCs and hESCs have been foundational to the generation of disease-relevant neural models, but as undifferentiated cells they do not model multicellular interactions or neural circuitry. Stem cell-derived neural progeny and 3D model systems help address some of these limitations. For example, HD iPSC-derived MSNs exhibit impaired viability and function with decreased spontaneous neuronal firing when compared to control MSNs. 13 These mutant neurons also show reduced sodium currents and altered calcium signaling, emphasizing intrinsic deficits in excitability and synaptic function. 46 In comparison, organoid models provide spatial organization to examine early circuit development. HD cortical organoids display premature or disorganized subcortical projections during corticogenesis, mirroring alterations in neuronal circuitry displayed in HD pathogenesis. 33 Assembloids further permit the examination of longer-range interactions and neuronal projections. In Str-SN assembloids, HD MSNs recapitulated the results of 2D models showing lower sodium currents and enhanced calcium activity compared to controls. 17 This model also exhibited deficits in axonal projections between regions, mimicking HD-related circuit disintegration. 17 Together, these findings demonstrate that while 2D cultures can reveal intrinsic neuronal impairments, 3D organoids and assembloids more reliably capture the multicellular architecture and connectivity disruptions characteristic of HD.
Modeling neurodevelopmental defects in vitro
Evidence from postmortem brain tissue and human stem cell-derived models suggests that the HD disease process includes a neurodevelopmental component. HD brain tissue exhibits higher rates (6.4–8.2x) of developmental defects. 12 Other structural anomalies visualized in human and mouse HD brains include defects in neural cell polarity and differentiation, alterations in cell cycle progression, and abnormalities in migration of progenitor cells.7,47 Given that the striatum is among the most vulnerable brain regions in HD, it was a major breakthrough for disease modeling when HD iPSCs and hESCs were successfully differentiated into GABAergic striatal neurons, permitting mechanistic and therapeutic studies.21,41 These models have enabled investigation into how HTT mutations disrupt early neurodevelopmental processes. Knockout of HTT disrupts specification of early and committed neural stem cells, causing impairments in the formation of ectodermal and mesodermal layers 48 as well as delayed neural induction. 49 iPSCs harboring mHTT display abnormal transitions out of the pluripotent state, including disrupted OCT4 expression and impaired coordination of early differentiation. 50 Once differentiation initiates, HD hESCs display erroneous neural fate specification, with precocious formation of NPCs and bias towards glial lineages, likely due to dysregulated Notch and Hes5 signaling. 49 Notably, mHTT also appears to impair medial ganglionic eminence (MGE) interneuron differentiation from NPCs, which may be significant given the essential role of MGE-derived GABAergic interneurons in neural circuit function and their involvement in neuropsychiatric disorders. 51
Although these findings emphasize developmental perturbations, the aging dimension of HD is a critical aspect of the disease that remains poorly understood despite extensive investigation. Prolonged culture and exposure to metabolic or oxidative stressors have all been used to model late-onset features and age-related pathologies. It is plausible that such cellular defects accumulate or are compensated for until a pathological threshold is reached, at which point cellular resilience mechanisms fail. Identifying the molecular pathways that sustain compensation, including enhanced proteostasis, adaptive stress signaling, or synaptic remodeling, could uncover novel therapeutic avenues through which to prolong neuronal function and delay disease onset.
Regional fate defects are difficult to capture in 2D cultures. In contrast, organoids form 3D structures that develop region-specific cell types and architecture, enabling the study of how mHTT disrupts tissue-level neurodevelopment and cell-cell interactions. Because so much has recently been learned from organoids in this regard, we dedicate an entire section to it below.
Recent findings using brain organoids
This section highlights the use of organoids and assembloids to model HD, focusing on neurodevelopmental abnormalities and transcriptional changes observed in these model systems. It covers defects in cortical layering and changes in organoid size linked to impaired neuroepithelial organization. We also emphasize the value of assembloids for studying interregional brain interactions and address ongoing efforts to refine organoid models to both understand HD pathogenesis and identify potential therapeutic targets.
In addition to mirroring neurodevelopmental patterns of the human fetus, organoids have complex organ-like spatial cell arrangements that foster both paracrine and direct contact-mediated interactions.32,52 Compared to stem cells and their neural progeny, brain organoids possess two features that may offer further insight into pathogenic mechanisms. First, because they consist of differentiated cells, the enhanced quality control mechanisms reported in stem cells are likely absent.22,53 Second, their adaptability to extended culture durations (e.g., four months 9 ) enables them to mimic neurodevelopmental stages and provides sufficient time for early disease-associated characteristics to emerge. Although organoids cannot replicate the age-dependent features of HD or other neurodegenerative disorders, their multicellular composition and long-term viability in culture enable investigations of early disease-associated characteristics and their variation across cell types.
Recent research suggests that altered brain development sets the stage for neurodegeneration during adulthood.1,7 Given that brain organoids recapitulate aspects of neurodevelopment, researchers have explored whether this model system mirrors the aberrant developmental patterns reported in HD fetal brains.9,15–17,33,34,43,50 Specifically, investigators have sought to determine if organoids similarly exhibit: (1) altered distributions of tight and adherens junction proteins9,15,16,33; (2) reduced numbers of mitotic progenitor cells9,16,17,33,43; (3) changes in the quantity and morphology of cilia at the apical surface9,33,34; and (4) an increased ratio of intermediate progenitor cells to apical radial glia in the ventricular zone of the developing cortex. 33 In general, organoid and assembloid studies mimic the altered neurodevelopmental patterns observed in HD fetal brains.7,9,15–17,33,34,43,50 With respect to organoid size, the literature is inconsistent on whether HD organoids are larger or smaller than their wild-type counterparts.9,16,33,34,43 Of the five studies available for review, three reported that HD organoids are typically smaller.9,16,33 This size reduction likely stems from the presence of smaller neural rosettes, indicating a reduced apical radial glia population and impaired neuroepithelial organization.15,16,33,43,50
Close comparison of the transcriptional profiles of HD versus wild-type organoids has provided insights into the mechanistic underpinnings of early neurodevelopmental changes. In HD organoids, upregulated genes are primarily associated with the following: neurodevelopment, epigenetic regulation, cytoskeletal organization, extracellular matrix organization, cell cycle regulation, stress response, and protein translation/degradation.9,16,33,34,43,50 In contrast, downregulated genes are largely linked to the following: later stages of neurodevelopment (e.g., neuronal and glial maturation and migration), ventral telencephalon development, interneuron specification, cytoskeletal organization and motility, cell cycle regulation, and neurotransmission and metabolic signaling—particularly within GABAergic and glutamatergic pathways.9,15,16,33,34,43,50 Many differentially expressed genes overlap with those identified in postmortem brain tissue from HD patients, suggesting that their dysregulation is an early event and persists throughout the patient's life.9,15 This sustained dysregulation may contribute to ongoing neurodegeneration and symptom progression, highlighting potential targets for therapeutic intervention across different stages of the disease.
Collectively, these transcriptional changes likely contribute to a developmental shift in HD organoids involving premature neurogenesis and delayed maturation of the resulting postmitotic neurons.16,33,34,43 In the developing cerebral cortex, neurons migrate along apical radial glial scaffolds to the cortical plate, where they are sequentially deposited in an inside-out pattern to form the six-layered cortical structure. Early born neurons populate the deeper layers (layers 6 and 5), while later-born neurons contribute to the upper layers (layers 4-2). As they migrate toward their final destinations, neurons transiently express markers specific to the cortical layers they traverse.54–56 Expanding on this in the context of HD pathogenesis, Liu et al. observed an accumulation of cortical layer 5 neurons (CTIP2+) in HD cortical organoids, accompanied by a reduction in upper-layer neurons (SATB2+, layers 4-2). 33 The accumulation of layer 5 neurons in HD organoids suggests that upper-layer neurons fail to complete their migration. 33 To assess whether this developmental shift renders HD organoids ‘older’ or ‘younger’ relative to their wild-type counterparts, many studies have analyzed organoid transcriptional profiles alongside those of human fetal brains. To date, these findings are mixed: some studies suggest HD organoids are ‘older’, while others suggest they are ‘younger’.33,43,50 Given the role of mitochondria in cellular aging, these shifts in developmental timing may relate partially to mHTT-induced mitochondrial dysfunction. Organoid studies show that mHTT disrupts mitochondrial dynamics, reduces oxidative phosphorylation, and contributes to neurodegeneration.16,57 These findings align with broader mitochondrial impairments in HD, including deficits in energy metabolism and neuronal signaling that are critical for cognitive function and synaptic plasticity.
In summary, the HD organoid field remains in its early stages, with much still to be uncovered. Scientists are actively refining organoid generation protocols and beginning to elucidate the cellular and molecular mechanisms underlying HD pathogenesis. Below is a summary of key organoid studies from the past year (2024–2025).
Traditionally, organoid research has relied on models of single brain regions. Liu et al. employed HD cortical organoids as a spatiotemporal model to characterize neurodevelopmental defects. 33 Organoids carrying CAG repeat expansions in the adult-onset range (55 or 59 repeats) had disrupted junctional complexes in neural tubes, delayed maturation of postmitotic neurons, aberrant specification of cortical neuron subtypes, and abnormal subcortical projections during corticogenesis. Regardless of disease status, the organoids contained polyQ assemblies associated with Golgi stacks and clathrin-positive vesicles; however, the structural and functional characteristics of these assemblies differed between genotypes. In HD organoids, the polyQ assemblies were shorter and scaffolded fewer Golgi stacks and clathrin-positive vesicles, which coincided with reduced ADP-ribosylation factor 1 (ARF1) recruitment to these assemblies. Pharmacological inhibition of ARF1 in wild-type organoids reproduced the junctional complex defects present in HD organoids, implicating ARF1-mediated Golgi dysfunction as a downstream consequence of mHTT. Together, these findings link polyQ pathology to disrupted cellular organization during cortical development and provide mechanistic insight into how mHTT impairs corticogenesis. 33 Lisowski et al. added to the growing body of HD research by generating isogenic iPSC lines with 70 CAG/CAA repeats in one or both HTT alleles, as well as a line in which the repeat expansion was eliminated from both alleles. 16 They conducted a comparative analysis of iPSCs, NPCs, and cerebral, cortical, and midbrain organoids derived from these lines. This approach led them to identify coiled-coil-helix-coiled-coil-helix domain containing 2 (CHCHD2)-mediated neurometabolic failure as an early feature of HD pathogenesis. 16
As another example of brain region-specific modeling, Galimberti et al. generated telencephalic organoids that include both dorsal and ventral cell fates. 9 During human neurodevelopment, the telencephalon gives rise to the cerebral cortex and basal ganglia, including the striatum. Because the cerebral cortex and striatum are the two most vulnerable brain regions in HD, this model holds promise for uncovering pathogenic mechanisms. The telencephalic organoids were derived from a panel of isogenic hESC lines with varying CAG repeat expansion lengths (20, 48, 56, or 72 repeats). HD organoids displayed abnormalities in ventral fate acquisition, resulting in fewer GABAergic neurons. In contrast, dorsal cell populations were less affected and primarily showed defects in maturation. 9
In HD patients, selective regional vulnerability and SRI lead to a mosaic of repeat expansion lengths across brain cells. To investigate the role of this mosaicism in HD pathogenesis, Galimberti et al. generated organoids composed of both mutant and wild-type cells. Compared to monoculture HD organoids, many abnormalities were rescued in the mosaic organoids. These findings suggest wild-type cells exert a beneficial non-cell-autonomous effect, which could be leveraged for therapeutic development. 9
To capture interregional interactions and cell diversity, the organoid field has increasingly prioritized the production and examination of assembloids. Notably, Wu et al. were the first to develop a protocol for generating HD organoids that exhibit mHTT aggregates rather than assemblies. 17 This protocol involves fusing striatum and midbrain substantia nigra (SN) organoids to form Str-SN assembloids. As this model mimics other disease-associated characteristics, including impairments in neuronal projections and decreased calcium signaling, it holds promise as a platform to study disease progression and test potential therapeutic strategies. 17 Świtońska-Kurkowska et al. were the first investigators to generate brain organoids modeling juvenile-onset HD that are larger than their wild-type counterparts. 34 This may be due to their use of fused dorsal-ventral assembloids, which mimic interactions between the developing cortex and striatum. These HD assembloids had enlarged neural rosettes and impaired neurogenesis, marked by delayed neuronal maturation and elevated expression of proliferation-related genes. While the impaired neurogenesis aligns with previous findings from cerebral and cortical organoids,16,33,43 the presence of enlarged neural rosettes contrasts with earlier reports.15,16,33,43,50 In addition to having altered proportions of inhibitory neurons and glia, HD assembloids also exhibited increased abundance of choroid plexus cells. These cells secrete transthyretin (TTR) into the cerebrospinal fluid, from which it may enter the bloodstream—highlighting TTR as a potential blood-based biomarker for HD. 34
Comparison of the ease and utility of various stem cell-derived model systems
Table 1 summarizes and compares features of stem cell models. iPSCs and hESCs provide genetically defined, indefinitely self-renewing platforms for modeling HD. While it can be challenging to create isogenic iPSC and hESC lines, doing so is critically important as isogenic controls eliminate potentially spurious differences elicited by genetic changes outside of the disease mutation. Stem cells require careful maintenance to prevent unwanted differentiation, yet their capacity to differentiate into any cell type makes them a powerful tool for studying disease mechanisms. While they can model transcriptional dysregulation and mimic early HD neurodevelopmental stages, they are limited in their ability to replicate cellular circuitry. Moreover, they only fully exhibit aberrant HTT behavior or SRI when proteasomal stressors or hyper-expanded HTT are present—a limitation likely due to the enhanced quality control mechanisms present in stem cells.22,53
Comparison of 2D and 3D stem cell-based models for HD research.

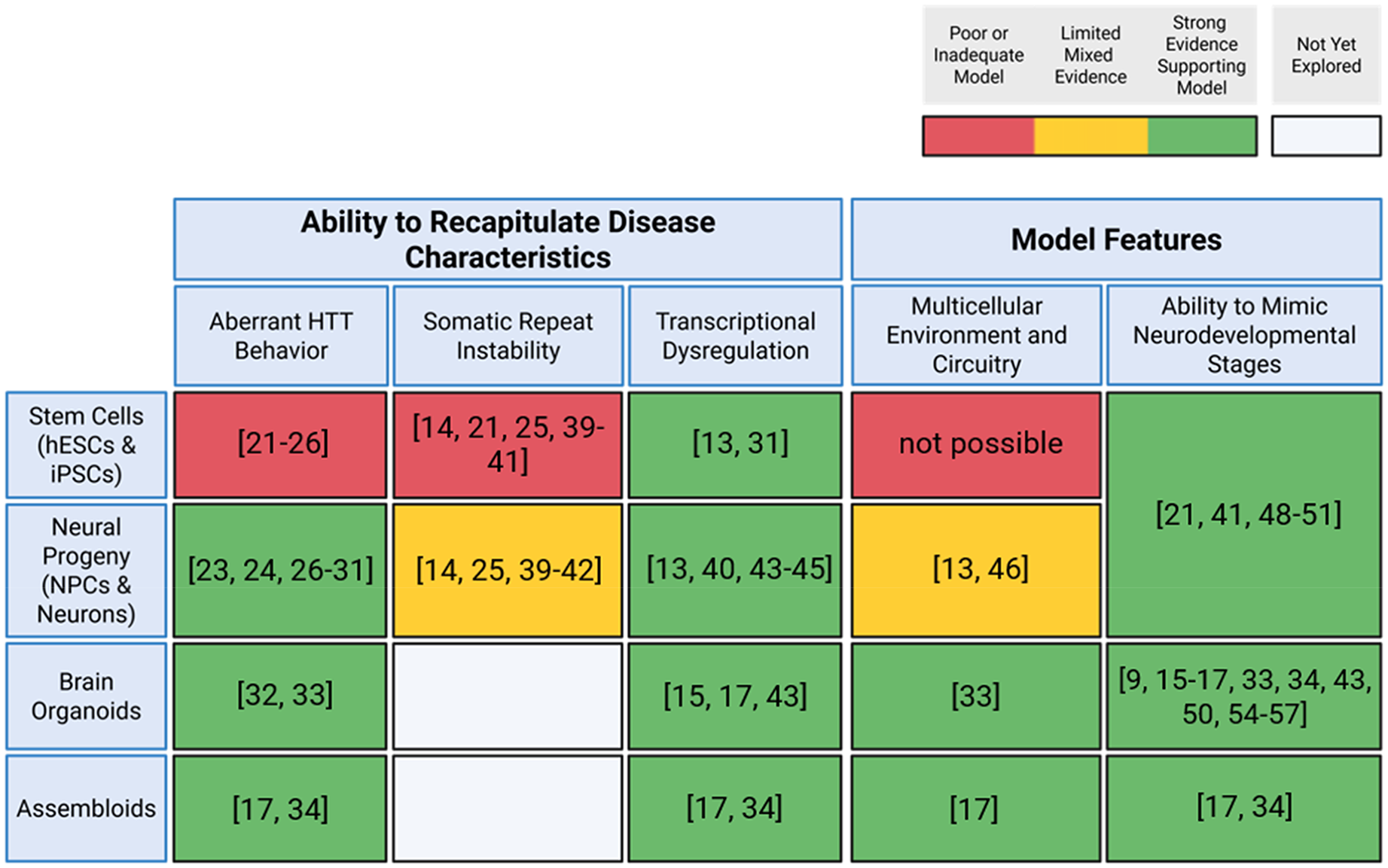
This table summarizes the ability of human stem cell-derived systems to recapitulate key disease-associated characteristics of HD. Created with BioRender.com.
NPCs, which can be generated from pluripotent stem cells in approximately 3–4 weeks, 58 require less maintenance than their undifferentiated counterparts, making them attractive for functional assays and drug screening. Differentiated neurons can model aberrant HTT behavior, SRI, transcriptional dysregulation, cellular circuitry, and early neurodevelopmental stages, making them a compelling option for modeling disease pathology. The challenge of maintaining neurons for extended periods, however, may prevent certain disease-associated characteristics from being captured. Moreover, some features may only manifest in the context of complex, multicellular environments.
Brain organoids provide greater structural and cellular complexity but are technically demanding due to variability in their maturation. They often require 1–2 months of culture to develop meaningful architecture and HD-relevant phenotypes. Organoids permit broader modeling of aberrant HTT behavior, cellular circuitry, transcriptional dysregulation, and neurodevelopmental changes across different areas of the brain. Unguided differentiation relies on the intrinsic capacity of pluripotent stem cells to self-pattern and produce diverse brain-like regions, offering heterogeneity and the potential to model interregional interactions. 59 These benefits also make the organoids highly variable, raising concerns about reproducibility. In contrast, direct differentiation relies on applying defined differentiation factors to guide organoid development towards specific regional identities, such as dorsal forebrain or midbrain. This ensures greater reproducibility and regional precision while at the same time sacrificing diversity and spontaneous organization that may reveal emergent developmental phenotypes.
Assembloids augment the ability of organoids to model circuitry, but are the most labor intensive, requiring the coordinated fusion and maintenance of two or more region-specific organoids. While offering invaluable information, their reproducibility and scalability remain limited.
Two-dimensional iPSC, NPC, and neuronal cultures primarily capture cell-autonomous processes, such as alterations in gene regulation and proteostasis that are intrinsic to affected cells. In contrast, organoids and assembloids facilitate investigation of non-cell-autonomous mechanisms, including interactions between neuronal subtypes, between neurons and glia, 60 or between different brain regions. Overall, NPCs offer the balance of speed and accessibility, while organoids and assembloids sacrifice ease for greater neurodevelopmental and in vivo disease relevance.
Advanced computational and transcriptomic methods are opening new doors in organoid research:
We conclude by highlighting the still unrealized potential of organoids. Incorporation of artificial intelligence (AI) and deep learning strategies into brain organoid research is likely to transform the analysis of neurodevelopmental and neurodegenerative processes. For example, AI-based analysis resulted in the development of a high-throughput, micropatterned, brain organoid-based platform for drug screening. 61 This platform employs deep convolutional neural networks (CNNs), which extract features that distinguish HD from non-HD organoid images, such as the expansion of neuroepithelial cores. The AI-based system detected subtle rescue effects evoked by bromodomain inhibitors on neurodevelopmental abnormalities in HD organoids. 61 The potential of AI to quantify both the toxic and beneficial effects of pharmacological treatments could be invaluable to the drug discovery process.
In addition to evaluating phenotypic rescue, AI can be used to analyze organoid structure and development by enhancing high-resolution imaging modalities and facilitating the interpretation of complex datasets. An interesting example is an AI-optimized segmentation and analysis pipeline that combines 3D volumetric magnetic resonance imaging (MRI) data with deep learning algorithms to produce 3D masks of organoids and segment aberrant features, such as cysts. 62 This pipeline enables longitudinal monitoring of cerebral organoid growth and, as a result, supports automated assessment of organoid quality, making it easier to track structural changes over time. 62 The integration of AI with advanced imaging analysis has the potential to uncover disease-associated characteristics that are often overlooked in 2D and 3D model systems.
Bulk and single-cell RNA sequencing (RNA-seq) have revolutionized biomedical research. Both approaches, however, require tissue dissociation, hindering their ability to assess spatial changes and cell-cell interactions. To address this limitation, researchers used MRI data from pre-manifestation HD patients alongside postmortem single-nucleus RNA-seq data to correlate cell loss with dysregulated genes, including those involved in development. 3 More recently, spatial transcriptomics have provided high-resolution gene expression maps of intact brain tissue and organoid sections, preserving their spatial organization. In HD mice, spatial transcriptomics revealed regional and age-dependent changes. These changes were detected at birth, with the gene expression profile at postnatal day 0 (P0) displaying a compensatory activated state among many cell types that shifted towards downregulation by 4 weeks of age. Using gene expression maps, the authors assessed regional vulnerability within the brain. They found heightened glycolysis in the caudate putamen as early as P0, alterations to MSN identity and maturation, and enrichment of astrocytes in the ventricular zone. 63 Single-cell transcriptomics has also revealed changes in glia, astrocytes, and striatal cell states as well as compensatory mechanisms,64–66 matching some of the results gleaned using spatial transcriptomic data.
Likewise, organoid morphology inferred by spatial transcriptomics can be used to determine when and how cellular and tissue architecture deviates from normal.67,68 For example, in HD midbrain organoids, single-cell gene expression maps have revealed upregulation of genes related to synaptic signaling, axon guidance, and neurodevelopmental defects that can be aided by spatial mapping to delineate where changes occur in relation to organoid structure and infer cell-cell interactions. 16 Rather than simply examining the gene expression map of an organoid section, spatial transcriptomics can help construct 3D molecular maps of organoids using serial sections and probe-based arrays. 69 Overall, spatial transcriptomics is already being used in HD mouse models and to visualize brain organoid transcription profiles, so utilizing it in HD organoids may further our understanding of developmental alterations resulting from the disease.
Beyond advances in AI and transcriptomics, organoid intelligence-based biocomputing represents an exciting frontier. 70 Recent research demonstrates that brain organoids harbor the essential molecular machinery and functional properties required for learning and memory, including synaptic plasticity, expression of immediate early genes, and highly interconnected, maturing neural networks bearing signatures of criticality. Furthermore, high-density microelectrode array recordings and stimulation of highly patterned electrical activity demonstrate that organoids are capable of both spontaneous and evoked, input-specific forms of plasticity. These advances speak to their potential as dynamic in vitro model systems to characterize disease-related network dysfunction. 71 Researchers have expanded organoid computing reservoirs by creating paired systems in which two organoids establish functional network connections. Experiments using virtual environments with reward and penalty stimuli demonstrate that these paired organoid systems can learn to “navigate” a digital maze. 72 Paired organoid reservoirs could be integrated into drug-discovery platforms to assess drug-induced changes in cognition, which cannot be assessed using 2D systems, and bypass blood-brain barrier permeability issues encountered in in vivo systems. In the study of HD, for example, high-density microelectrode array recordings from disease and non-disease organoids could serve as functional read-outs for changes in organoid network formation.
Together, these developments highlight the convergence of biological and computational sciences, revealing novel opportunities to understand and treat HD. The dual functionality of organoids as both disease models and biocomputing units underscores their potential as powerful tools with which to study disease mechanisms and advance therapeutic discovery. By bringing together AI, spatial transcriptomics, and biocomputing, researchers may soon achieve fundamental advances in our understanding and treatment of neurodevelopmental disturbances in HD and other neurological disorders.
Footnotes
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Stem cell work and the writing of this review were supported by NINDS R35NS122302 (HP), NIH MSTP T32 T32GM007863 (NG), NIH Cellular and Molecular Biology Training Grant T32-GM007315 (RP).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.