
Abstract
Background
Huntington's disease (HD) is a neurodegenerative disorder caused characterized by motor, cognitive, and psychiatric/behavioral impairments. Among these symptoms, irritability is particularly burdensome due to its frequency and significant impact on the daily lives of both patients and caregivers. Currently, no FDA-approved medication specifically targets this symptom.
Objective
To assess the efficacy of dextromethorphan/quinidine (DM/Q) 20/10 mg (NUEDEXTA®) in managing irritability in HD.
Methods
Double-blind, placebo-controlled, randomized, crossover clinical trial. Participants underwent a 13-week study: six weeks of DM/Q treatment, one-week washout, and six weeks of placebo. The DM/Q 20/10 mg regimen started with a once-daily dose, increased to twice daily after the first week, and tapered back to once daily in the sixth week. Participants were evaluated during three in-person visits: at baseline and at the end of each treatment phase. Assessments included motor and cognitive exams, the Irritability Scale, and the Problem Behavior Assessment–short version (PBA-s) (ClinicalTrials.gov NCT03854019).
Results
Twenty participants were enrolled, with 18 completing the study (mean age = 43.94 ± 10.70 years, 11 females and 7 males). Both DM/Q and placebo reduced mean Irritability Scale scores (32% and 27.5%, respectively) and the irritability subscale of the PBA-s (42% and 33%, respectively), with no statistically significant differences between groups. Additionally, DM/Q showed no significant advantage over placebo in motor, behavioral, or cognitive outcomes.
Conclusion
DM/Q did not demonstrate significant benefits in managing irritability compared to placebo. Further research with larger sample sizes and alternative therapeutic strategies is needed to effectively address this symptom in HD.
Plain language summary
Huntington's disease is a neurological disorder that affects movement, thinking, and mood. One particularly challenging symptom is irritability, which can impact both individuals with the disease and their caregivers. Currently, there are no approved medications specifically designed to treat irritability in Huntington's disease. To address this, we designed a pilot study to investigate whether a drug called dextromethorphan/quinidine (DM/Q), also known as NUEDEXTA®, could help reduce irritability in people with Huntington's disease. Over the course of 13 weeks, 18 participants took part in a trial where they received both the actual medication and a placebo (a dummy pill) in alternating order, without knowing which was which. The results indicated that both the actual drug and the placebo reduced irritability to some extent, but there was no significant difference between the two. Additionally, the drug did not show any additional benefits compared to the placebo regarding movement, thinking, or behavior. In summary, this study found that DM/Q was not more effective than a placebo in treating irritability in Huntington's disease. Further research is needed to identify effective treatments for this symptom.
Introduction
Huntington's Disease (HD) is a neurodegenerative disease caused by an abnormal expansion of CAG trinucleotide repeats in the Huntingtin gene (HTT) on chromosome 4. While HD is well-known for its characteristic abnormal movements, especially chorea, psychiatric and/or behavioral symptoms may manifest years before the onset of motor symptoms and can negatively impact quality of life and functional independence of individuals living with HD. 1 Among several neuropsychiatric symptoms, irritability and emotional dysregulation are common in HD gene expansion carriers (HDGECs). These behaviors are a matter of significant concern for their family members, who frequently bear the related impact. 2 Irritability is characterized as a mood state that includes feelings of frustration and anger, making the affected individual more likely to respond strongly to external stressors.3,4 Although irritability usually manifests through behaviors such as verbal outbursts or aggression, this is not always the case. The internal frustration experienced by irritable individuals may not always be evident through external behaviors.3,5 Consequently, irritability can place a significant emotional burden on both individuals and their families, potentially jeopardizing the daily functioning of those affected and complicating or hindering the care provided by their families.
Multiple medications have been used to treat irritability with varying degrees of success. These include selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitor (SNRIs), tricyclic antidepressants (TCAs), antipsychotics, buspirone, benzodiazepines, mood stabilizers, and beta-blockers.6,7 However, there are currently no pharmacological therapies approved by the U.S. Food and Drug Administration (FDA) for treating irritability in HD. A potentially promising option is NUEDEXTA®, a combination of 20 mg dextromethorphan hydrobromide and 10 mg quinidine sulfate (DM/Q 20/10 mg), which is a FDA-approved drug for the treatment of pseudobulbar affect. 8 Pseudobulbar affect is characterized by uncontrollable and inappropriate episodes of crying and/or laughing that occur in individuals with certain neurological conditions. The potential emotion-regulating effects of DM/Q have prompted investigation into its use for managing irritability and aggression in conditions such as Alzheimer's disease and autism.9,10 However, the findings have been mixed, with benefits observed in some behavioral domains but not others. For instance, in individuals with autism spectrum disorder, DM/Q was linked to reductions in irritability, yet no significant changes were reported in aggression scores. 9 Conversely, in Alzheimer's disease patients exhibiting clinically significant agitation, DM/Q significantly decreased agitation and aggression scores, while the irritability/lability domain showed no meaningful improvement compared with placebo. 10 In the absence of an FDA-approved medication for treating irritability in HD, this double-blind, randomized clinical trial was designed to evaluate the efficacy of DM/Q as a potential treatment option for HDGECs who are experiencing irritability.
Methods
Study design
The study was funded by Cures Within Reach and conducted at The University of Texas Health Science Center at Houston (UTHealth Houston). The study received approval from the local institutional review board (HSC-MS-18-1049). It was also registered on the online database of clinical research studies, ClinicalTrials.gov (NCT03854019). Signed consent forms were obtained from each individual. This research adhered to the U.S. Code of Federal Regulations, FDA guidelines, and Good Clinical Practice quality standards. Additionally, this article follows the Consolidated Standards of Reporting Trials (CONSORT) recommendations for reporting the findings.
In a double-blind, randomized, placebo-controlled, crossover trial, participants received DM/Q 20/10 mg, marketed under the brand name NUEDEXTA®, and placebo. The 13-week crossover design included six weeks of treatment for each arm, separated by a one-week washout period. During the six-week treatment period, participants received DM/Q once daily for the first week (adaptation period), DM/Q twice daily for the following four weeks, and finally, DM/Q once daily for the last week (tapering period) (Figure 1).
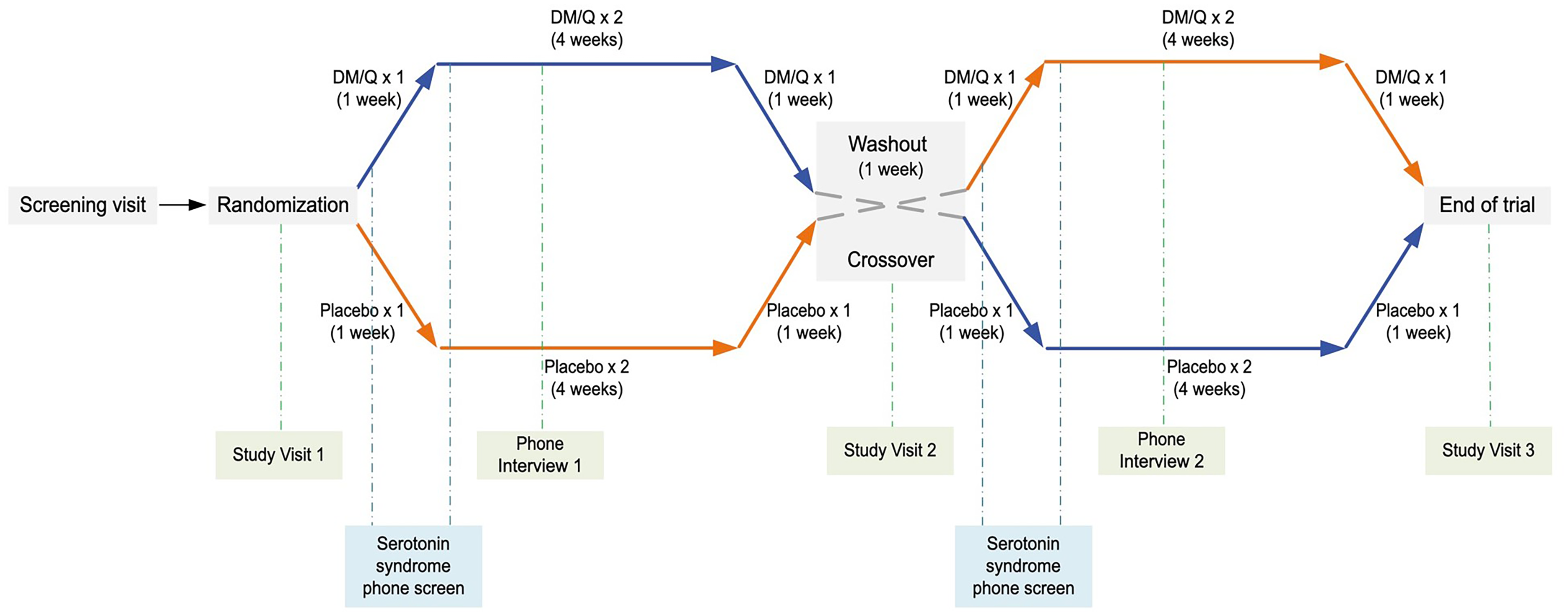
The study's crossover design. The DM/Q pill contained 20 mg of dextromethorphan and 10 mg of quinidine. Each arm included an adaptation period (1 pill/day for 1 week), a main treatment period (2 pills/day for 4 weeks), and a tapering period (1 pill/day for 1 week).
Participants & eligibility criteria
We screened HDGECs at the Huntington's Disease Society of America (HDSA) Center of Excellence at UTHealth Houston from August 2019 to November 2022. The following inclusion criteria were used for recruitment: (i) genetically verified HD mutation (CAG repeat > 36); (ii) age between 18 and 75 years; (iii) a score of 14 or higher on the self-report version of the Irritability Scale; 11 (iv) stable concomitant medications for at least 30 days prior to screening; and (v) capacity to make an informed decision.
The exclusion criteria included (i) known hypersensitivity to dextromethorphan or quinidine; (ii) pregnancy or breastfeeding; (iii) active suicidality as defined by the Columbia Suicide Severity Rating Scale (C-SSRS); 12 (iv) lack of a highly effective method of contraception in men and pre-menopausal women; (v) presence of any medically uncontrolled underlying disease (e.g., hypertension, diabetes mellitus); (vi) clinically significant renal failure (creatinine clearance < 30 ml/min) or hepatic dysfunction; (vii) specific history of heart issues (complete heart block, self or family history of QTc prolongation, torsades de pointes); (viii) history of unexplained syncope within the past year; (ix) current use of strong CYP3A4 inhibitors, first-generation antipsychotics, monoamine oxidase inhibitors, tricyclic antidepressants, or tetrabenazine; (x) presence or history of seizures or diagnosed epilepsy; (xi) cognitive impairment defined as a score < 18 on the Montreal Cognitive Assessment (MoCA); (xii) clinically relevant abnormal findings during the screening (physical exam, vital signs, ECG, and laboratory results, including complete blood count and metabolic panel); (xiii) parallel or recent participation (< 2 months) in another interventional trial; and (xiv) a high chance of non-compliance as determined by the Investigators.
Procedures & randomization
During the screening visit, participants provided consent, and eligibility was assessed based on the inclusion and exclusion criteria. Once eligibility was confirmed, participants were randomized to start the crossover trial with either DM/Q or placebo using a web-based program. Allocation was concealed using sequentially numbered drug containers with identical appearances (blinded capsules manufactured by Compounding Shop Pharmacy, Houston, TX). All participants and active research personnel, including the PI, were blinded to the treatment sequence and unmasking was maintained throughout the study period.
During the first arm of the trial, participants starting with either DM/Q or placebo were instructed to take one pill daily in week 1, two pills daily (one in the morning and one in the evening) from weeks 2 to 5, and one pill daily in week 6. A one-week washout period (week 7) separated the two arms of the study, during which participants did not receive DM/Q or placebo. During the second arm of the trial (weeks 8–13), participants crossed over to the alternate treatment: those who received placebo in the first arm received DM/Q in the second arm, and those who received DM/Q in the first arm received placebo in the second arm (Figure 1).
Assessments
Participants were assessed during three in-person visits: at baseline, during the washout period (week 7), and after the end of the second arm. To assess irritability, we used the Irritability Scale, which includes 14 questions about the presence of various irritable behaviors in the past two weeks. 11 The Unified Huntington Disease Rating Scale (UHDRS) motor, UHDRS diagnostic confidence level (DCL), and UHDRS total functional capacity (TFC) assessments were used to determine the motor status and functional stage of the disease. 13 The UHDRS motor ranges from 0 to 124, 0 being a lack of any abnormal movements. The DCL is a measure of certainty regarding the clinical diagnosis of HD based on motor signs. It ranges from 0 (normal) to 4 (motor abnormalities that are unequivocal signs of HD, with ≥99% confidence). 14 The TFC ranges from 0 to 13, with the stages of disease defined as follows: 11–13 is Stage I (least severe); 7–10 is Stage II; 3–6 is Stage III; 1–2 is Stage IV; and a score of 0 is Stage V (most severe). 15
Behavioral and/or neuropsychiatric symptoms of the participants were evaluated with the Hospital Anxiety and Depression Scale (HADS), Neuropsychiatric Inventory Questionnaire (NPI-Q), and Problem Behavior Assessment short version (PBA-s). HADS is a self-report questionnaire consisting of 7 items each for anxiety and depression (score range 0–21 for each subscale). 16 The NPI-Q evaluates 12 behavioral issues in patients with dementia and provides two scores, one for severity (score range: 0–36) and another for caregiver distress (score range: 0–60). 17 The PBA-s is a semi-structured clinical interview that measures the severity and frequency of 11 key behavioral symptoms in HD.18,19 The PBA-s items combine into five subscales: depression (depressed mood + suicidal ideation + anxiety), irritability (irritability + aggressive behavior), apathy (apathy), executive function (perseveration + obsessive-compulsive behavior), and psychosis (delusions + hallucinations). All behavioral rating scales were administered to participants with concurrent caregiver input, providing additional corroboration and improving the reliability of symptom ratings.
To evaluate global cognitive function, we used the MoCA, which ranges from 0 to 30, with higher scores indicating better cognitive performance. 20 Overall patient condition was assessed with Clinical Global Impression Severity (CGI-S) and Clinical Global Impression Improvement (CGI-I). 21 CGI-S ranges from “normal” (score 1) to “amongst the most extremely ill patients” (score 7) and CGI-I ranges from “very much improved” (score 1) to “very much worse” (score 7).
Safety measures
During the three in-person visits, participants were monitored for safety and adverse events through vital signs assessment, physical examinations, ECG, complete blood count, metabolic panel, and a rapid qualitative pregnancy test (for female participants). Suicidal ideation was also assessed using the C-SSRS at each visit. Additionally, we conducted two phone interviews midway through the treatment in each arm to screen participants for possible adverse events and medication compliance. For participants at risk of serotonin syndrome, additional phone screenings were conducted 24 h after the initial dose and 24 h after the first change to a twice-daily dosage in each study arm (Figure 1).
Statistical analysis
Statistical analyses were conducted using SPSS version 29 and R version 4.3.1. Group differences at the baseline visit were assessed using independent bivariate analyses with consideration of the treatment sequence (Mann-Whitney U test and Fisher's exact test). For the analysis of the crossover data, we employed paired bivariate methods, the Wilcoxon signed-rank test for continuous outcomes and McNemar's test for categorical outcomes. To model treatment effects, period effects, and treatment sequence effects, we utilized multivariable linear mixed-effects regressions. This approach was applied to a 2 × 2 crossover design, where all subjects are randomly assigned to one of two sequences, each receiving two treatments in succession, and the order of treatments is determined by the assigned sequence. The models were adjusted for age and sex (except for CGI-I and CGI-S). All statistical tests were two-sided and an alpha level of p < 0.05 was set as the level of statistical significance.
Results
A total of 20 participants were initially enrolled from a pool of 112 screened HDGECs. However, one participant withdrew during the first week of the trial due to adverse events, including nausea, dizziness, and headaches. Another participant was lost to follow-up (inability to contact since the first attempt, i.e., phone interview 1). As a result, the final sample consisted of 18 HDGECs, as shown in Figure 2. The mean age of the participants was 43.94 years, with a standard deviation of 10.70 years. The group included 11 females and 7 males. The baseline characteristics of the study participants are summarized in Table 1.

CONSORT flow diagram of the study participants.
Characteristics of the study participants in two treatment sequences at baseline.

Values are mean ± standard deviation or number (percentage).
DM/Q, dextromethorphan/quinidine; HADS, Hospital Anxiety and Depression Scale; MoCA, Montreal Cognitive Assessment; NPI-Q, Neuropsychiatric Inventory Questionnaire; PBA-s, Problem Behaviours Assessment short version; UHDRS, Unified Huntington Disease Rating Scale; UHDRS TFC, total functional capacity.
a. Mann-Whitney U-test.
b. Fisher's exact test.
The results of treatment with DM/Q and placebo are displayed in Table 2. Both DM/Q and placebo led to a reduction in the mean scores on the Irritability Scale by 32% and 27.5%, respectively (Figure 3A). Similarly, the irritability subscale of the PBA-s showed a 42% decrease for DM/Q and a 33% decrease for the placebo treatment (Figure 3B). Statistical analysis indicated that DM/Q and placebo did not differ significantly, as shown by the Irritability Scale (P = 0.71) and PBA-s irritability (P = 0.95) scores. After adjusting for potential confounders using linear mixed models, the results remained consistent, showing no significant differences between DM/Q and placebo observed for the Irritability Scale (P = 0.72) and the PBA-s irritability subscale (P = 0.98).
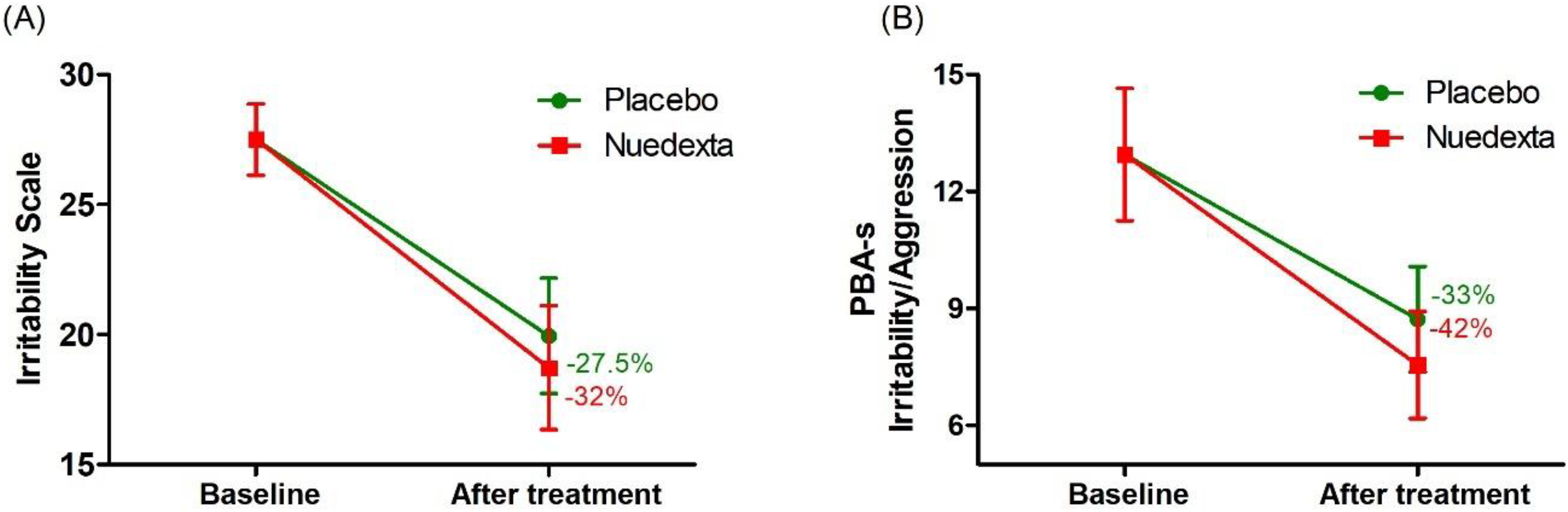
The effects of DM/Q and placebo on irritability compared to baseline (n = 18), as assessed by the irritability scale (a) and the PBA-s irritability subscale (b).
Outcomes with DM/Q treatment compared to placebo.
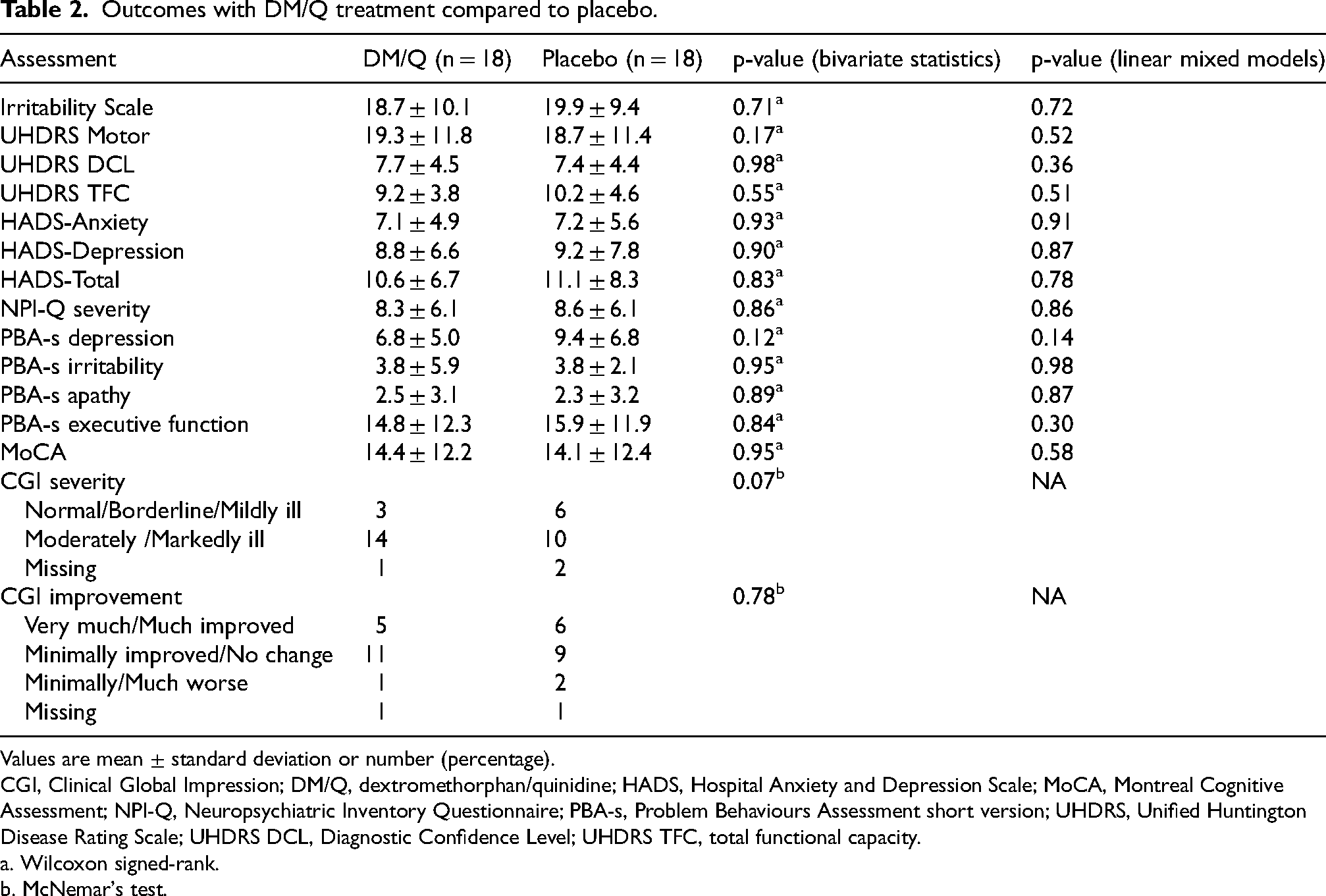
Values are mean ± standard deviation or number (percentage).
CGI, Clinical Global Impression; DM/Q, dextromethorphan/quinidine; HADS, Hospital Anxiety and Depression Scale; MoCA, Montreal Cognitive Assessment; NPI-Q, Neuropsychiatric Inventory Questionnaire; PBA-s, Problem Behaviours Assessment short version; UHDRS, Unified Huntington Disease Rating Scale; UHDRS DCL, Diagnostic Confidence Level; UHDRS TFC, total functional capacity.
a. Wilcoxon signed-rank.
b. McNemar's test.
Both DM/Q and placebo had similar outcomes in motor and functional assessments. No significant differences were found between DM/Q and placebo for the UHDRS motor score (P = 0. 17), UHDRS DCL (P = 0.98), and UHDRS TFC (P = 0.55). In behavioral and/or neuropsychiatric assessments, including the HADS (P = 0.83), NPI-Q (P = 0. 86), and various PBA-s subscales such as depression (P = 0.12), apathy (P = 0.89), and executive function (P = 0.84), DM/Q did not demonstrate any significant effects compared to placebo. Additionally, no effect of DM/Q was observed on the MoCA (P = 0.95) or on the general clinical status of participants, as measured by CGI severity (P = 0.07) and CGI improvement (P = 0.78). The results remained consistent when potential confounders were accounted for using linear mixed models.
Seven of the 20 participants reported adverse events, which included headaches, diarrhea, nausea, dizziness, and changes in appetite, libido, and mood. A detailed summary of adverse events by treatment arm is presented in Table 3.
Adverse events reported during the study, stratified by DM/Q and placebo arms.

Discussion
This double-blind, randomized, crossover clinical trial aimed to evaluate the efficacy of DM/Q in treating irritability in HD, addressing the lack of an FDA-approved medication for this condition. The results showed no statistically significant differences in the Irritability Scale and the irritability subscale of the PBA-s between DM/Q at a dosage of 20/10 mg taken twice daily and placebo. Additionally, DM/Q did not demonstrate significant effects in motor, behavioral or cognitive assessments.
Dextromethorphan, which has traditionally been used as an over-the-counter cough suppressant, has a complex pharmacological profile. It acts as an N-methyl-D-aspartate (NMDA) receptor antagonist, a sigma-1 receptor agonist, an α3β4 nicotinic acetylcholine receptor antagonist, and a weak serotonin and norepinephrine reuptake inhibitor.22,23 When taken independently, dextromethorphan undergoes rapid first-pass metabolism in the liver, leading to low and variable bioavailability. However, when co-administered with quinidine—a potent inhibitor of the cytochrome P450 2D6 (CYP2D6), enzyme responsible for metabolizing dextromethorphan—this process is blocked. As a result, quinidine enhances dextromethorphan's bioavailability, allowing higher concentrations to reach the central nervous system. 22 Given its complex neuropharmacological effects, the DM/Q combination has been investigated as a potential treatment for various neuropsychiatric or behavioral syndromes associated with neurological conditions, including agitation, irritability, and neurobehavioral symptoms in autism, and received FDA approval for the treatment of pseudobulbar affect.9,1024–28
Excitatory glutamatergic signaling, mediated through NMDA receptors, plays a significant role in emotional dysregulation. 29 The NMDA receptor antagonist effects of DM/Q can reduce aggression and irritability by modulating excitatory neurotransmission and stabilizing emotional regulation. 29 A randomized, placebo-controlled clinical trial by Chez et al. assessed the effects of DM/Q on irritability and aggression in 14 adults with autism. 9 The DM/Q regimen was initiated with a daily dosage of 20/10 mg, which was increased to twice daily after 7 days, provided the medication was well tolerated. 9 Both DM/Q and placebo resulted in reduced mean irritability scores on the Aberrant Behavioral Checklist; however, DM/Q showed a greater reduction, with a nearly 7-point decrease compared to a 3-point decrease with placebo, which was statistically significant. No statistically significant difference was found between DM/Q and placebo in the aggression scores, as assessed by the Overt Aggression Scale. 9 Cummings et al. evaluated the effect of DM/Q on 220 patients with probable Alzheimer's disease and clinically significant agitation. 10 Dosages were initially adjusted to 20/10 mg twice daily, with further increases to 30/10 mg twice daily during the trial. 10 This randomized, multicenter, double-blind, placebo-controlled study demonstrated a significant reduction in the NPI agitation/aggression domain among participants treated with DM/Q compared to placebo. However, the NPI irritability/lability domain showed no significant difference between DM/Q and placebo over the 10-week parallel-group analysis, with a mean change of −2.4 in the DM/Q group versus −1.8 in the placebo group. 10 A statistical difference between placebo and DM/Q in the irritability/lability domain emerged only in a secondary analysis, when the results were analyzed using a two-stage sequential parallel comparison design. 10 This design was used to enhance the ability to detect a true treatment effect, even when a strong placebo response is present. 30
In another study, Woodard et al. conducted a double-blind, placebo-controlled trial investigating the effects of dextromethorphan in eight children with autism and related developmental disorders who displayed increased irritability at baseline, as measured by the Aberrant Behavior Checklist. 31 Dextromethorphan hydrobromide syrup was administered at 30 mg every 12 h for children aged 6–12 and 60 mg every 12 h for those aged 12 and older. The study found no significant effect of dextromethorphan compared to placebo in treating irritability or core symptoms. 31 Only three of the eight participants demonstrated a 25% or greater improvement on the irritability subscale attributed to the medication. 31
Despite previous evidence supporting the efficacy of DM/Q in controlling irritability, 10 we observed no significant effect compared to placebo in reducing irritability in HD when administered at the maximum dose of 20/10 mg twice daily. Notably, both DM/Q and placebo reduced irritability scores by more than 25% from baseline, suggesting a strong placebo effect that may have masked the treatment effect of DM/Q, similar to findings by Cummings et al. 10
The placebo response is influenced by participants’ perceptions, expectations, and personality characteristics. 32 This effect is particularly challenging in studies involving behavioral and/or psychiatric symptoms, where outcomes rely heavily on self-reports and clinical observations, making them more susceptible to placebo-related biases. 30 Moreover, controlled study environments with regular follow-up care can amplify contextual effects—such as the study setting, participant expectations, and therapeutic interactions—potentially enhancing the placebo response. In clinical research, treatment efficacy is typically assessed by the net benefit, which represents the difference in improvements between the placebo and treatment groups. However, focusing solely on this difference may overestimate the clinical significance of the placebo response and underestimate the true overall treatment effect experienced by participants. 33 Additionally, the relatively short study duration (4 weeks of full-dose DM/Q treatment) may have limited our ability to detect sustained treatment effects. A longer study period could offer a more comprehensive assessment of DM/Q's efficacy in reducing irritability.
DM/Q was well-tolerated among our patients. A key safety consideration with the quinidine component of DM/Q is QT interval prolongation, 34 a condition that can increase the risk of serious arrhythmias, such as torsades de pointes. Therefore, a careful monitoring, particularly in patients with pre-existing cardiac conditions, is warranted. However, the current DM/Q dose, which contains 10 mg of quinidine, is significantly lower than the doses typically associated with a heightened risk of QT prolongation. 22 The risk of QT prolongation at this dose is considered minimal, although monitoring is still recommended for vulnerable populations. In our study, regular ECG monitoring was conducted, and no major cardiac side effects were observed. Similarly, an open-label, multicenter study of DM/Q in 553 patients with pseudobulbar affect across more than 30 neurological conditions reported no severe cardiac events. 35 The most common side effects in that study were nausea (11.8%), dizziness (10.5%), headache (9.9%), somnolence (7.2%), fatigue (7.1%), diarrhea (6.5%), and dry mouth (5.1%). 35 In our trial, the most frequently reported side effect attributable to DM/Q was headache, experienced by two participants. Other side effects were reported in only one instance or not at all.
This study has several limitations. The strong placebo effect, small sample size, and short treatment duration may have reduced the statistical power required to detect the true effects of DM/Q. Additionally, the wide variability in HD progression and symptomatology may have contributed to heterogeneous treatment responses. The inclusion of participants at different stages of the disease could have further influenced the outcomes and limited the generalizability of the findings. It is also important to acknowledge the limitations of the terminology and assessment tools used to measure emotional dysregulation. Traditional terms like irritability or outbursts may not fully capture the complexity of the symptoms/behaviors, as emotional dysregulation often involves a disproportionate response to minimal triggers, with patients describing rapid emotional escalation unrelated to baseline irritability. Additionally, the strong placebo response may partly result from the act of discussing this often-embarrassing symptom, providing participants with a sense of validation and relief.
In conclusion, irritability is a common and burdensome psychiatric symptom in HDGECs, often emerging years before the onset of motor symptoms and placing significant emotional and social strain on individuals and their families. While various off-label treatments are used to manage irritability in HD, there are currently no FDA-approved or globally accepted medications for this purpose. Our study did not demonstrate a significant benefit of DM/Q compared to placebo for irritability in HDGECs. This lack of observed effect may be partly due to the strong placebo response, which can obscure the true therapeutic impact of DM/Q. Further research with larger sample sizes and alternative therapeutic approaches are warranted to effectively address irritability in this population.
Footnotes
Acknowledgements
The authors sincerely thank all participants and their families for invaluable contributions to this study.
Ethical considerations
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. The study was approved by the UTHealth Houston Committee for the Protection of Human Subjects (HSC-MS-18-1049).
Consent to participate
Written informed consent was obtained from all individual participants included in the study. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was funded by Cures Within Reach.
Declaration of conflicting interest
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. In the past 12 months, EFS has received honoraria as an advisory board member, consulted for, and received research funding from CHDI Foundation, Huntington's Disease Society of America, Huntington Study Group/Neurocrine Biosciences, Prilenia, PTC Therapeutics, University of Iowa/NIH, UniQure, Teva Pharmaceuticals, Sage Pharmaceuticals, Wave Life Sciences, MedPage, and The Michael J Fox Foundation. NPR received research funds from the Diana & Conrad Weil, Jr. Family Foundation, the Huntington's Disease Society of America, the Alzheimer's Association, and the Texas Alzheimer's Research and Care Consortium. ALT has received honoraria as an advisory board member, consulted for, and received research funding from Bristol Myers Squibb, Lundbeck, National Institute on Aging, and the Texas Alzheimer's Research and Care Consortium.
Data availability
The datasets generated during and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request.
Author roles
1. Research project: A. Conception, B. Organization, C. Execution;
2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique;
3. Manuscript Preparation: A. Writing of the first draft, B. Review and Critique;
SAZ: 1B, 1C, 2C, 3A;
OC: 1B, 1C;
NK: 1B, 1C;
JP: 1B, 1C;
DD: 2A, 2B;
BD: 1B, 1C;
ALT: 3B;
NPR: 1A, 1B, 1C, 2C, 3B
EFS: 1A, 1C, 3B
All authors read and approved the final manuscript.