
Abstract
One of the great mysteries of Huntington disease (HD) is its adult onset. HD is caused by a CAG repeat expansion in the HTT gene, which encodes a protein (HTT) that is crucial for intracellular transport and neuronal growth, so one would expect development to be altered in some way. Human fetal samples, cell and animal model studies have shown that neurodevelopment is indeed altered in HD. Structural and functional abnormalities can be detected during the presymptomatic phase, and mutation carriers may display subtle cognitive deficits, psychiatric disturbances, or motor impairments years before clinical diagnosis. However, despite these early developmental alterations and the lifelong presence of mutant huntingtin (mHTT), HD carriers typically experience a long asymptomatic period. In this review, we provide an overview of the functions of HTT and the dysfunctions caused by mHTT or HTT loss during development, from gastrulation to neurulation, and examine the specific contribution of the cerebral cortex in HD. Drawing on nearly 25 years of research, we integrate studies ranging from in vitro to in vivo studies, highlighting the molecular mechanisms that contribute to HD neuropathology. Understanding these early developmental processes may define a window for therapeutic intervention, a period of plasticity during which the brain may still correct or compensate for developmental errors. This could strengthen the foundations to extend the presymptomatic phase and delay neurodegeneration.
Keywords
Introduction
Huntington's disease (HD) is an adult-onset neurological disorder caused by a CAG expansion in exon 1 of the Huntingtin (HTT) gene. The CAG repeats in HTT encode a polymorphic polyglutamine (polyQ) stretch in the N-terminal domain of the Huntingtin (HTT) protein. In the non-HD population, the CAG sequence is repeated 9 to 35 times. A CAG expansion exceeding 35 repeats results in HD. HD has classically been regarded as a progressive neurodegenerative disease characterized by selective vulnerability of the striatum and cortex, leading to motor, cognitive, and psychiatric manifestations. The motor and cognitive impairments in HD typically manifest in adulthood, but longitudinal studies have demonstrated that individuals carrying the HTT mutation display subtle but measurable abnormalities decades before the clinical onset of the disease.1,2 These early manifestations encompass cognitive, psychiatric, and functional changes, accompanied by alterations to brain morphology, connectivity, and fine motor performance.3,4
Historically, the movement disorder of HD, caused by a dysfunction and degeneration of the striatum, was considered to be its most characteristic feature. Consequently, the study of striatal degeneration received the most emphasis, while the contribution of the cerebral cortex to HD has been largely neglected. Without underestimating the autonomous role of striatal dysfunction, there is growing evidence to suggest that striatal degeneration is caused at least partly by defective cortical signaling to the striatum.5–8 The disruption of cortical signaling may interfere with the correct maturation of striatal neurons. Studies in mouse models of HD and cells derived from patients have revealed defects in the proliferation, differentiation and maturation of striatal medium spiny neurons.9,10 Moreover, the genetic manipulation of presynaptic compartments by microfluidic approaches has demonstrated that the nature of the presynaptic neurons governs the functionality of the whole cortico-striatal network. 6 Wild-type cortical neurons are sufficient to rescue network functionality and survival signaling in the HD striatum. Conversely, HD cortical neurons are sufficient to impair striatal signaling and the overall functioning of the cortico-striatal network. Zhao and coworkers have also shown in vivo that an enhancement of the cortical trafficking of BDNF is sufficient to rescue striatal atrophy. 8 This suggests that, in HD, the cortical defects actively drive and modulate HD progression rather than simply reflecting the downstream consequences of striatal degeneration.
The cortex is altered early in the disease, as demonstrated by multiple neuroimaging studies showing global cortical and subcortical atrophy in HD mutation carriers’ decades before the clinical onset of disease. 11 Abnormalities develop as early as during embryogenesis, as fetuses carrying even the mildest HTT expansions, which would be predicted to cause late-onset symptoms, present cortical abnormalities by 13 weeks of gestation. 12 In mouse models, both wild-type and mutant huntingtin (mHTT) play crucial roles in pre- and postnatal development. HTT is involved in cortical progenitor cell division, neuronal migration, and dendritic and glutamatergic neuron maturation, suggesting a crucial role in cortical development.12–18 In HD, these functions are altered, leading to abnormal cortical development and contributing to late-onset disease.
Here, we describe HD longitudinally along the cortical developmental axis, from pluripotent stem cells to neural stem cells and mature neurons. We discuss the early cellular and molecular events contributing to developmental defects and disease onset.
The role of HTT begins during gastrulation
Embryogenesis starts with a single fertilized egg that undergoes successive rapid divisions. These divisions produce a solid ball of 100 cells called a morula, in which the cells are tightly packed and largely undifferentiated. As the morula develops, it undergoes cavitation to form a blastula (an unpolarized spheroid of polarized epithelial cells) and then a gastrula with a well-established body axis (anteroposterior and dorsoventral). Gastrulation is characterized by large-scale cell movements and significant morphogenetic changes to establish the three primary germ layers: ectoderm, mesoderm, and endoderm. In mammals, gastrulation-associated signaling begins post-implantation, following invagination of the primitive streak.19,20 Human HTT mRNA and protein are detectable before implantation and persist throughout gastrulation. 21 Studies on HD during gastrulation are lacking, even though HTT appears to play a crucial role at this stage (Figure 1A). In mouse, complete HTT knockout during gastrulation is embryo-lethal (causing death between embryonic day 7.5, E7.5 and E8.5).22,23 Mice with the deletion of a single HTT allele are viable but display motor and cognitive deficits as adults. 24 By contrast, the expression of low levels of mHTT in a knockout background lead to perinatal death,25,26 revealing the importance of gene dosage during development. Consistent with these findings, heterozygous variants of HTT are associated with an autosomal recessive neurodevelopmental disorder. 27
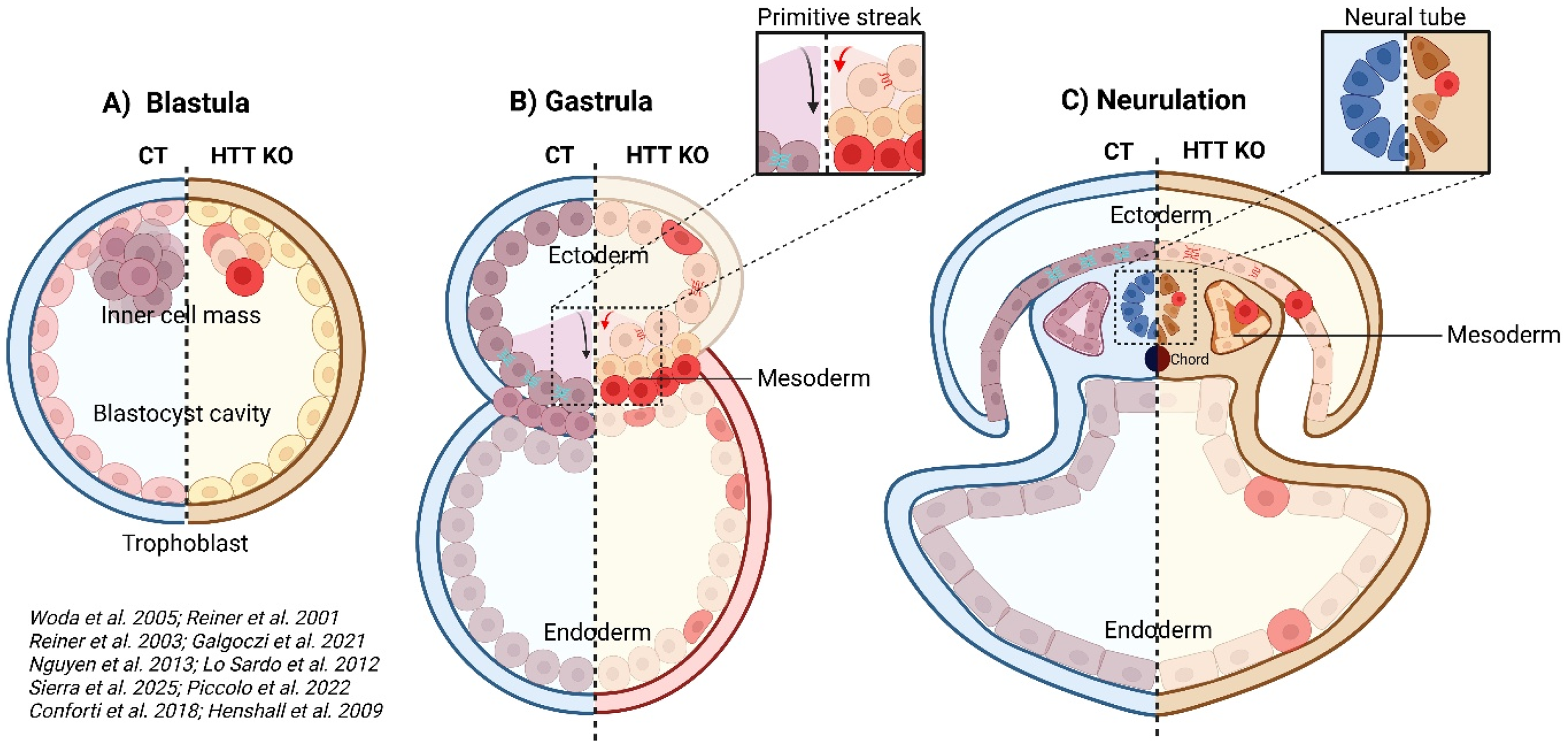
HTT functions begin during gastrulation. (A) HTT KO blastula shows abnormal cell differentiation and death. (B) During gastrulation in an HTT KO environment, junctional alterations guide cells to differentiate preferentially towards the mesoderm at the expense of the ectoderm, thereby reducing the pool of neuronal precursors. The primitive streak is also shortened, leading to defects in the anterior/posterior axis. (C) Cellular adhesion and differentiation are impaired during neurulation in the germ layers and neural tube, which is disorganized and contains aberrant cell types. Red: less differentiated cells, CT (blue part): control, HTT KO (orange part): HTT knock-out. Printed with permission from BioRender.
Detailed analyses of E7.5 HTT-depleted embryos have revealed early patterning defects in the formation of the anterior/posterior axis, marked by a shortened primitive streak and smaller paraxial mesoderm due to a weaker or mislocalized expression of differentiation factors (Figure 1B). 28 Head development is also altered by the lack of Otx2 expression. HTT levels are crucial for the correct execution of morphogenetic programs during gastrulation, probably because they coordinate the expression of key morphogens during early tissue patterning. Moreover, chimeric blastocysts composed of a mixture of wild-type and HTT-depleted embryonic stem cells can develop normally.29,30 However, HTT-depleted embryonic stem cells display lower levels of differentiation and have higher cell death rates in cortical and striatal areas. By contrast, when normal embryonic stem cells are introduced into mouse blastocysts completely devoid of HTT, the embryos do not survive.29,30 HTT is, therefore, required to ensure the differentiation and survival of neural progenitors in mouse, for cortical and striatal regions.
The ectoderm (outer layer) of the gastrula gives rise to the brain and spinal cord; the mesoderm (middle layer) gives rise to muscles, bones, and blood, and the endoderm (inner layer) develops into the gut, liver, and lungs. The generation of these layers is tightly controlled to ensure correct tissue formation but this control is disrupted by both pathological CAG expansions in the HTT gene or a loss of HTT. In mouse embryonic stem cells and in human gastruloids lacking functional HTT, cells preferentially differentiate into mesoderm cells at the expense of ectoderm cells, potentially decreasing the pool of neuronal precursors.31,32 The mechanism involved is linked to disrupted cell polarity: HTT mutations or depletion compromise tight junctions, leading to a mislocalization of apical proteins, and alterations of the orientation and organization of epithelial cells crucial for gastrulation. These defects also interfere with key developmental signaling pathways, including the TGFβ, BMP, and Notch pathways, which normally guide cell-fate decisions. Evidence from HTT-null embryonic stem cells and zebrafish embryos suggests that HTT is essential for homotypic interactions in neuroepithelial cells, affecting neurulation and rosette formation, 33 highlighting the importance of HTT during early tissue organization and for the establishment of polarity.
Finally, the polyQ tract itself is biologically active in neural progenitors and plays an important role in rosette formation and cellular polarity, as its deletion or reduced length disturbs the spatial organization of progenitors inside the rosettes. 34
Abnormal neurulation and cortical development with consequences during adulthood
Neurulation emerges directly from the gastrulation blueprint, transforming the newly established ectodermal layer into the neural foundation of the central nervous system. Occurring during the third to fourth week of human gestation, neurulation involves precise coordination of cell shape changes, planar cell polarity, and morphogen signaling to establish dorsal-ventral and anterior-posterior patterning of the future brain and spinal cord (Figure 1C). The specification and patterning of the neural ectoderm is controlled by a conserved network of BMP, Wnt, FGF, and Shh signaling.
FGF signaling support neural induction alongside BMP inhibition and posteriorize the neural plate, establishing anterior-posterior identity. Wnt signaling (particularly Wnt/β-catenin) caudalizes the neural plate, promoting hindbrain/spinal cord identity over forebrain fates, while Wnt/PCP pathway activity drives convergent extension movements essential for neural plate folding and tube closure.
Sonic Hedgehog (Shh) from the notochord patterns the ventral neural tube (floor plate, motor neurons) during and after neurulation. BMP4 inhibition is the primary trigger for neural induction; high BMP4 promotes epidermal (non-neural) fate, while organizer-secreted antagonists (Chordin, Noggin, Follistatin) block BMP4 to enable neural ectoderm identity. The cellular response to BMP4 gradients is modulated by activation of Yes-associated protein (YAP), which is essential for spatial partitioning of the embryonic ectoderm. Recent human embryonic stem cell (hESC) ‘neuruloid’ models have begun to recapitulate these early patterning events in vitro, offering a window into how mHTT might disrupt the earliest stages of neural tube formation. 35 In wild-type neuruloids, YAP activity is tightly regulated by mechanical cues and morphogen signaling at early stages (day 4); low-level YAP activation sensitizes cells to BMP4 stimuli, promoting non-neuronal ectoderm induction. One study reports that mHTT induces ectopic YAP activation at day 4 of neurulation, disrupting ectodermal lineage specification by skewing differentiation toward epidermis at the expense of neural crest. 36 Once the neural plate is specified, apical constriction and PCP-dependent convergent extension drive folding and fusion of neural folds to generate the neural tube, after which brain regionalization proceeds with the emergence of the three primary brain vesicles at the anterior end. In HTT-depleted neural cells derived from human embryonic stem cells or cells carrying CAG expansions, mitotic spindle orientation becomes randomized, impairing the formation of organized neuronal rosettes. 37 The disruption of mitosis in multipotent progenitors leads to the generation of aberrant multinucleated cells, neurons with enlarged somas, and DNA strand bouquets; it is associated with abnormal centrosomes and aneuploidy. In human HD induced pluripotent stem cells, neuronal differentiation is impaired from the early neuroectodermal stage, interfering with the acquisition of both striatal and cortical neuron identities. 38 This defect is associated with misexpression of the pluripotency marker OCT4 and the neuroectodermal determinant PAX6, resulting in immature, disorganized rosettes of progenitors with unspecified cortical and striatal fates. Similarly, in zebrafish, HTT is essential for correct neural plate. 39
Once neurulation is completed, the cortex, which is organized into six distinct layers, begins to develop from the neuroepithelium lining the ventricular zone containing neural progenitors, such as radial glial cells and intermediate progenitor cells. Radial glial cells, the primary neural stem cells in the ventricular zone, can divide symmetrically to expand the progenitor pool or asymmetrically to produce one progenitor and one neuron. Intermediate progenitor cells, derived from radial glia in the subventricular zone, undergo a limited number of divisions and primarily generate neurons, contributing directly to cortical neurogenesis. The newly generated neurons then migrate radially to form the layered cerebral cortex in an inside-out pattern. This developmental process spans embryonic days 10 to 17 in mice 40 and gestational weeks 7 to 18 in humans. 41 Over the last decade, HTT has been shown to play key roles in the developing mouse cortex in the divisions of neuronal progenitors, the migration of newly generated neurons, and the maturation of their somato-dendritic compartment (Figure 2).
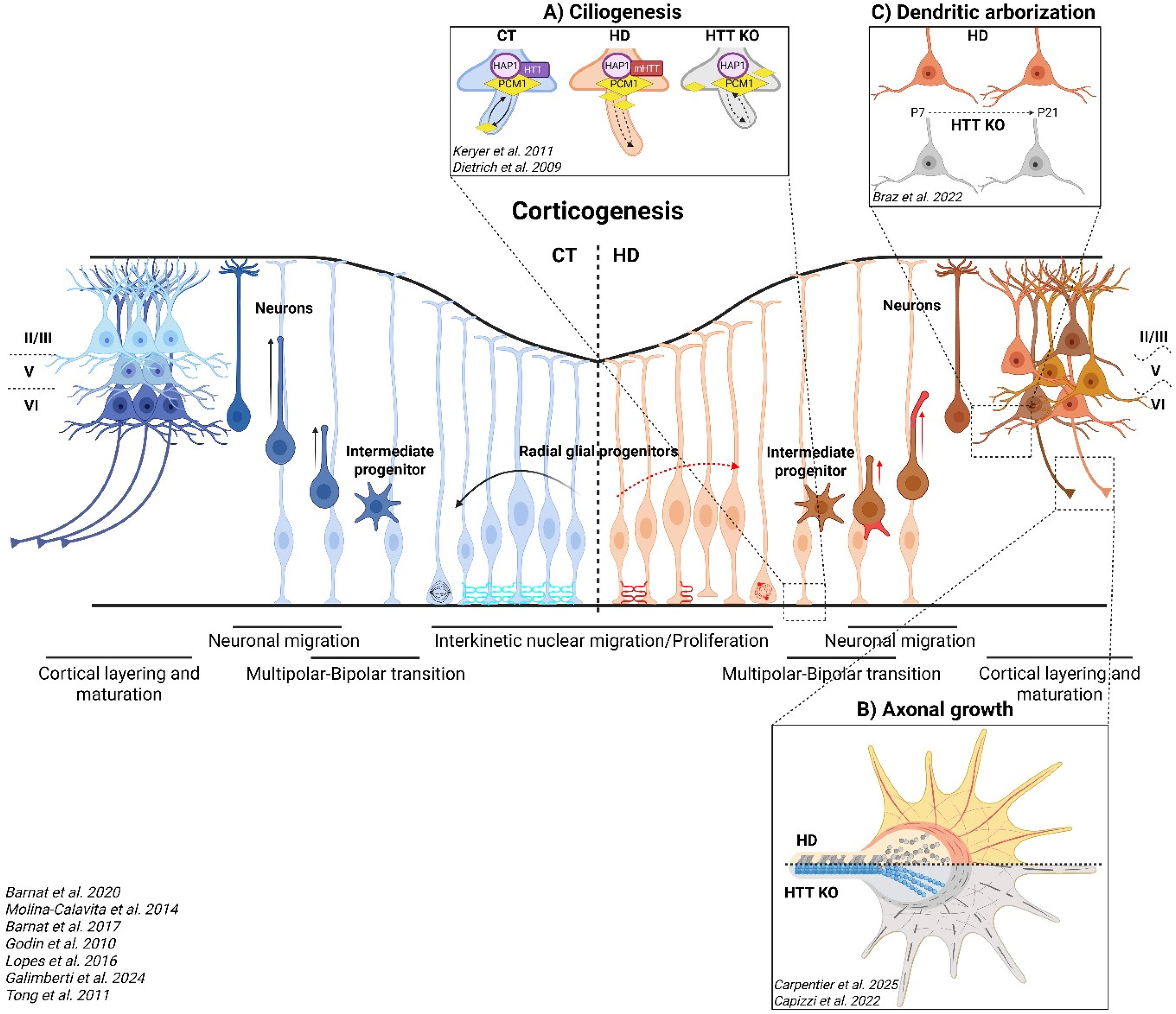
Abnormal cortical development due to HD or HTT depletion. Every step of corticogenesis is altered in HD and HTT depletion from the cell cycle of radial glial progenitors to the migration and maturation of newly generated neurons. (A) Primary cilia are misorganized and are either increased (HD) or reduced (HTT-KO) in length in radial glial progenitors. (B) Microtubule and actin defects in the growth cones of HD and HTT KO neurons, respectively. (C) Increased or decreased dendritic arborization of post-natal HD and HTT KO neurons, respectively. CT (blue part): control, HD (orange part), HTT KO (gray part): HTT knock-out, P7: post-natal day 7, P21: post-natal day 21. Printed with permission from BioRender.
HTT controls the maintenance of stemness and the generation of cortical neurons through its role throughout the cell cycle in radial glia cells, from interphase to mitosis. During interphase, the interkinetic nuclear migration of mouse HD progenitors is impaired and nuclear movements are slower. 12 HTT localizes to the spindle pole of mitotic cells and both HTT inactivation and the expression of mutant forms of HTT lead to misorientation of the spindle via changes in dynein, NUMA1, and the p150(Glued) subunit of dynactin, affecting cell fate.15,42,43 Similarly, HD telencephalic organoids are smaller than control ones and contain a smaller pool of polarized progenitors that are less engaged in mitosis, with fewer mitotic cells and a higher cell cycle exit ratio. 44
The alteration of cell cycle progression in radial glia cells is correlated with an accumulation of HTT protein at the apical endfeet in human HD carrier fetuses at gestation week 13 and HD mice, whereas this protein spreads throughout the entire ventricular zone in control conditions. 12 Moreover, the neuroepithelial barrier created by tight and adherens junctions is disrupted in human HD fetuses and mouse HD embryos by the mislocalization of ZO1, PAR3, β-catenin and N-cadherin (Figure 2). Neural epithelial structures in human HD cortical organoids are also altered due to deficiencies in junctional complexes. Central to this mechanism are long, oriented polyQ assemblies that associate with Golgi stacks in neural progenitor—structures essential for establishing apical-basal polarity and directing vesicular trafficking during neural development. Contrary to conventional views of aggregates as purely pathological, these endogenous polyQ assemblies appear physiological and protective, stabilizing Golgi architecture through recruitment of ADP-ribosylation factor 1 (ARF1), a small GTPase involved in membrane trafficking. 45 Incorporation of mHTT into these structures compromises their integrity, resulting in approximately 40% reduced ARF1 recruitment. Consequently, this impedes formation of clathrin-mediated vesicles, directly obstructing the transport of junctional proteins to the cell membrane. The ARF1-dependent defect propagates to disrupt epithelial integrity, altering spatiotemporal control of corticogenesis. Critically, pharmacological restoration of ARF1 recruitment rescues junctional complex formation and normalizes progenitor layering. During corticogenesis, the disruption of adherens junctions causes radial glial cells to lose their apical endfeet, leading to their detachment from the neuroepithelium. Loss of radial glial contact with the ventricular surface impairs sensing of cilium signaling and disrupts proper segregation of cell fate determinants during divisions.
HTT dysfunction also directly impacts cilium biology through impaired PCM1 trafficking (Figure 2A). In HTT-deficient cells, failure to transport PCM1 to the pericentriolar material (PCM) produces short or absent cilia. Conversely, polyQ-expanded HTT blocks PCM1 removal from the PCM, causing its accumulation at the ciliary base and resulting in abnormally long cilia. 46 Both conditions yield dysfunctional cilia through distinct molecular mechanisms. This extends to motile cilia as well: HTT inactivation in Wnt1 lineages reduces ependymal cell ciliation in the midbrain, disrupting cerebrospinal fluid circulation and resulting in closure of the Sylvian aqueduct and development of hydrocephalus in mice model. 47
HTT is also required later in neuronal development, for the correct migration and maturation of the newly generated neurons. Neuronal maturation begins with a transition from multipolar to bipolar morphology, which is altered by HTT depletion. 17 The newly generated bipolar neurons then migrate through the neocortex toward the marginal zone by locomotion along radial glial fibers. The leading process, which develops into the apical dendrite, extends forward continually and wraps around radial glia, whereas the trailing process grows from the rear and becomes the future axon, extending toward the intermediate zone. HTT depletion results in defective cell migration, with fewer cells entering the cortical plate. 48 This is accompanied by an increase in the number of cells entering the cell death pathway. Moreover, as for HTT-depleted neurons, the presence of mHTT delays radial migration and affects the multipolar-bipolar transition of a subset of postmitotic neurons (Figure 2).17,48 The proposed mechanism involves the HTT- and Rab11-mediated transport of recycling endosomes containing N-cadherin adhesion molecules. HTT loss of function triggers a cytoplasmic and perinuclear accumulation of N-cadherin, altering the establishment of cell-to-cell adhesions in the leading process, these adhesions being required for the radial migration of newly generated neurons along glial fibers.
Cell migration and axonal navigation result from combined responses to guidance of molecules and physical barriers. For instance, the subarachnoid space of the meninges prevents cortical neurons from overmigrating into the marginal zone, ensuring that their cell bodies remain confined within the cortical plate. A gradient of Sema3A from the marginal zone simultaneously attracts apical dendrites while repelling axons. In the pyramidal neurons of mice with HD, apical dendrites are misoriented, suggesting defects of Sema3A signaling. 49 At the level of the axonal growth cone, HD or HTT-depleted neurons display microtubule and actin disorganization, impairing axonal growth (Figure 2B).18,50 While the absence of HTT triggers a reduction of axonal length due to actin unbundling, the polyQ expanded form of HTT binds and bundles F-actin cytoskeleton as the non-expanded one. 50 In contrast, in HD, the reduced axonal length results from the impairment of microtubules. 18 Cytoskeletal disorganization is accompanied by a decrease in FAT atypical cadherin 4 (FAT4), 18 potentially due to the higher levels of ADAM10 metalloprotease activity observed in HTT-depleted cells. 33 HD induced pluripotent cells-derived cortical neurons also have shorter neurites and delayed functional maturation, as demonstrated by their lower spontaneous and evoked electrophysiological activity. 51
HTT depletion or mutation during mouse development results in changes to cortical layering. 15 Similarly, human HD cortical organoids have fewer cortical projection neurons and altered neuronal layering associated with lower levels of calcium activity and an aberrant targeting of cortical projections to the striatum. 45 In mouse, excitatory HD or HTT-depleted neurons have abnormally low levels of synaptic transmission, enlarged spines, greater excitability, and shorter, less complex dendritic arbors (Figure 2C).14,16,52 These deficits become apparent during the first week after birth, but they self-correct in the second week in HD but not in HTT-depleted neurons, restoring the functioning of the circuit to wild-type levels. If these defects are corrected in the first week of life, when they first occur, the HD mice do not develop signs of HD or the disease itself later in life. Thus, this critical developmental window of HD-related alterations has long-term consequences for connectivity and network activity. 14 Indeed, the developmental expression of mHTT or the downregulation of wild-type HTT is sufficient to induce deficits in adulthood, including cortical and striatal abnormalities, axonal degeneration, and motor impairment.13,53
The structural differences during early neurodevelopment are supported by the antagonistic pleiotropy theory of HTT in HD. 54 The theory is based on the idea that some genes associated with disease, which have detrimental effects during ageing, could have beneficial effects at earlier stages. Indeed, a larger brain volume and cortical features such as larger surface area and greater folding were observed in premanifest carriers along with enhanced cognitive, behavioral and motor capacities, before a prolonged deterioration of functional and structural processes. 55 Interestingly, and in line with the well-established observation that cortical and striatal structures are profoundly affected in adulthood, the beneficial pleiotropic effect appears to be confined to cortical regions. Although the cortex initially exhibit an increased volume, this advantage progressively diminishes during the early stages of the disease. These findings are supported by a study involving young developing premanifest adults, where higher CAG repeats before the onset of neurodegeneration were associated with improved cognitive abilities. 56
Omics studies reveal an earlier but slower differentiation of HD cells
Cellular and molecular approaches have shed light on dysfunctions during HD cortical development, but little is known about the molecular changes occurring early on in development. Throughout development, molecular modifications are tightly and dynamically regulated, highlighting the specificity of HTT function and dysfunction at each stage of development. At the time point at which differentiation to neural fate occurs, HD modifies the expression of genes specific to cellular development mechanisms. 57 Ring et al. found that HD induced-pluripotent stem cells displayed no genetic modifications, but that the “neuronal development” and “dorsal striatum formation” biological function gene modules were disrupted in HD neural stem cells, in which the TGFβ and Netrin-1 genes were the most frequently modified. 57 By contrast, another study involving a single-cell analysis of human HD embryonic stem cells provided support for early transcriptional changes. 58 In pluripotent cells, the HD mutation has been shown to affect the levels of proteins from cell survival pathways. 59 Oxidative stress- and cytoskeleton-related proteins are disrupted in human HD embryonic stem cells, increasing sensitivity to cell toxicity. Together, these results demonstrate that mHTT affects each developmental stage in a specific manner. There also appears to be a certain cell-type specificity as the transcriptomic profiles of myotubes and hepatocytes differentiated from human HD embryonic stem cells do not differ from those of controls, whereas neural progenitor cells and neurons express HD-specific molecular phenotypes. 60
The non-pathogenic polyQ tract itself regulates early-to-late neural development genes in a Q-length dependent manner, with neuroepithelial organization and neuronal differentiation genes showing differential expression that supports the hypothesis that HTT polyQ length directly modulates neuronal differentiation. 34 In HD models, mHTT disrupts methylation regulators (Dnmt1 downregulation, Gadd45g upregulation) and transcription factor binding (SOX2, FRA-2/JUND), leading to altered DNA methylation and persistent silencing of neurogenesis genes (Sox2, Pax6, Nes) from ESC through post-mortem stages, a finding directly confirmed by ChIP-seq in STHdhQ111 cells. 61 This epigenetic predisposition intersects with stage-selective splicing dysregulation, where mHTT drives differentiation-stage-specific aberrant splicing in neuronal development, contributing to neurodevelopmental disorganization. 62 Some alternative splicing patterns observed in ESC models persist in post-mortem HD tissues, indicating that developmental splicing defects may contribute to pathogenesis later. 62 The dysregulation particularly affects splicing factor genes (e.g., SRSF2, PTBP2, TSEN54, TCERG1) from the earliest ESC stage, potentially creating a feedback loop where mis-splicing of the spliceosome machinery amplifies global splicing errors. Neuronal development genes like MACF1 (neuronal migration) and PTPRD (neurite outgrowth) show CAG-dependent splicing changes that disrupt neuronal connectivity, while mitochondrial regulators such as DNM1L/DRP1 link splicing dysfunction to metabolic vulnerability. Moreover, proteomic and metabolomic integration identifies CHCHD2 as a central hub whose dysregulation impairs mitochondrial complex IV assembly and activates mitochondrial integrated stress response (mISR) in NPCs, disrupting metabolic programming required for neural commitment. 63 Crucially, while mHTT broadly disrupts mRNA processing, its own splicing is aberrant, generating small N-terminal fragments that form nuclear inclusions, a hallmark of pathogenicity. 64
Multi-omic approaches have validated the phenotypes described at cellular level. As shown in the above-described studies of mouse models and human tissues, transcriptomic modifications reveal that the phenotype of HD cortex-derived cells is immature relative to that of controls. 51 Longitudinal studies have been performed on these cells. Initially (culture day 0, D0), HD iPSC-derived cortical cells display a deregulation of development and proliferation-related genes whereas, at D60, the “cellular morphology” and “neurological diseases” pathways are differentially expressed relative to control cells. At later stages, HD leads to changes in the expression of genes involved in cell adhesion, axon guidance, voltage-gated potassium channels and synaptic properties that are probably linked to an immature state. HD induced pluripotent stem cells-derived striatal neurons and astrocytes, and mouse astrocytes are also populated with immature cells displaying an enrichment in the expression of genes related to axon guidance adhesion molecules, extracellular matrix, immature astrocytes and mitosis, reflecting an alteration of neurodevelopment.65–69 Similarly, when considering late developmental stages, HD cortical 45 and telencephalic organoids 44 have transcriptomic signatures consistent with premature neurogenesis but delayed neuronal maturation in HD.
Interestingly, while most of the molecular modifications observed in post-mortem HD samples relate to neurodegeneration, abnormalities of the transcriptomic signatures of developmental pathways are also maintained at late stages of HD. A genome-wide analysis of mRNA levels in the prefrontal cortex revealed that 19% of the genes differentially expressed between controls and HD carriers were related to immune response, neuroinflammation and development. 70 Similar results were obtained for HD mice in a study in which a single-nucleus RNAseq analysis performed on the adult cortex and striatum revealed a decrease in the level of astrocytic marker gene expression, demonstrating immaturity. 69 What is the significance of the abnormal expression of development pathways in degenerating HD brains? As their name suggests, developmental pathways are highly active during development, but they are expressed throughout life, to maintain cellular homeostasis. For instance, the Wnt, Notch and Shh pathways are widely studied as key developmental pathways required for almost all aspects of neurogenesis, from the proliferation and survival of progenitors to their migration, neuronal synaptic plasticity and maintenance. 71 Given the complex crosstalk between these signaling mechanisms, they may modulate neurodegeneration in HD adult brains as suggested for other neurodegenerative diseases. Another possibility concerning the modified expression of neurodevelopmental pathways during neurodegeneration is that developmental mechanisms are reactivated to compensate for neurodegeneration, as during the regeneration observed followed a nerve lesion. 72
Discussion
One of the most striking features of HD is the long presymptomatic period, during which individuals carrying the CAG expansion remain clinically normal despite developmental changes and the lifelong presence of mHTT. This resilience may be due to compensatory mechanisms enabling the brain to buffer developmental changes. The human cortex develops over a remarkably extended period, from embryogenesis through adolescence, encompassing prolonged phases of neurogenesis, synaptic pruning, and plasticity. This extended developmental timeline provides multiple opportunities for adaptive remodeling, which may temporarily counteract the deleterious effects of mHTT.
Direct studies of the role of HTT in fertilization and morulation are lacking, but the available evidence suggests that HTT is essential from the earliest developmental stages. Correct doses of HTT appear to be indispensable for these morphogenetic programs: both the complete loss of HTT and excessive decreases in HTT levels are lethal, demonstrating the dosage sensitivity of this molecule. HTT may orchestrate morphogen gradients, establishing and maintaining embryonic polarity, and guiding early lineage specification. The establishment of polarity may be mediated by molecules interacting directly with HTT: the actin cytoskeleton and HTT-associated protein 40 (HAP40) adaptor protein.50,73 HAP40 is crucial for endosomal trafficking and may be involved in the RAB11-dependent trafficking of NCAD and the recycling of polarity complexes, such as PAR3 and aPKC complexes. 17 The ability of HTT to bind actin may contribute to cytoskeletal reinforcement and bundle reorganization in the establishment of polarity, as shown for growth cone remodeling during axonal development. 50
During embryonic corticogenesis, mHTT alters the balance between progenitor proliferation and differentiation, promoting premature neuronal differentiation at the expense of progenitor maintenance.15,42,43 This earlier differentiation may compromise later phases of cortical maturation, including synaptic refinement, pruning, and circuit remodeling. As in the musculoskeletal system, neuronal networks require repeated rounds of activity-dependent remodeling to achieve functional strength. When differentiation occurs prematurely, critical phases of synaptic regeneration and reinforcement may occur prematurely, resulting in connections that are initially functional but less resilient in the long term. This may render cortical circuits particularly vulnerable when somatic CAG repeat instability emerges later in life, triggering neurodegeneration.74,75 In mice, postnatal cortical development is perturbed by mHTT expression: axonal outgrowth and dendritic branching are initially reduced, delaying circuit establishment.14,18 However, between postnatal days 21 and 26, neurons in cortical layers II/III display a compensatory peak in dendritic arborization with a restoration of connectivity despite early deficits. 14
Neuroimaging studies in premanifest HD carriers have provided evidence for similar compensatory mechanisms in humans. The increase in activation in prefrontal and parietal regions during cognitive tasks suggests that network-level reorganization maintains function. 76 Such adaptive cortical recruitment may, however, come at a high price: excessive or inefficient activation can increase metabolic demand and accelerate neural fatigue, marking a transition from compensation to decompensation during the progression of HD. In addition, the superior early-life function driven by mHTT brain structural reorganization happen at the cost of later life neurodegeneration and decline, highlighting the antagonistic pleiotropy of HTT. 55 The HTT polyQ tract may have facilitated nervous system evolution, as humans possess the longest non-pathogenic CAG expansions, a feature potentially linked to cortical folding during hominid evolution.34,54,77 This evolutionary advantage carries a clinical burden, however: longitudinal data show that in carriers with CAG36-42 repeats, each additional CAG repeats correlate with subtle cognitive decline and accelerated striatal atrophy. 78 These observations suggest that the polyQ tract persists through a fragile equilibrium between requisite neurodevelopmental functions and mutational instability, rather than active selection for longer alleles.
Together, these findings support a model in which HTT is required for the coordination of early morphogenetic processes, the establishment of polarity, and neural progenitor survival. In HD, the coexistence of mHTT toxicity with low levels of wild-type HTT results in a delicate balance, allowing survival and development but building vulnerabilities into the cortical and striatal circuits. In early life, these defects are buffered by plasticity and redundancy, making it possible to maintain functioning at near-normal or even advantageous levels. However, as somatic CAG repeat instability increases during adulthood,74,75 antagonistic pleiotropy of HTT switch to the detrimental phase with aging, 55 and compensatory mechanisms decline, 79 the initially latent developmental irregularities may emerge as a neurodegenerative disease.
Recent HTT-lowering strategies hold promises, yet their implementation requires careful consideration of HTT dose-dependent effects. Current approaches (antisense oligonucleotides, RNAi, and base-editing) differentially spare HTT, yet none achieve complete selectivity. Encouragingly, the ongoing POINT-HD trial (NCT05677769) directly addresses this challenge by testing WVE-003, an allele-selective antisense oligonucleotide that specifically targets mHTT while preserving wild-type HTT through CAG-repeat-length-dependent chemistry. However, the consequent 50% reduction in wild-type HTT dosage raises concerns about whether residual levels can sustain neuroprotective functions throughout disease progression. While half HTT dosage is compatible with embryonic development, symptomatic neurons may have a heightened dependence on the neuroprotective functions of HTT, making adult-stage reduction particularly concerning. Few studies that have examined the long-term effects of further reducing HTT levels support this caution. Heterozygous HTT knockout mice exhibit impaired motor activity, cognitive deficits, and neurodegeneration in the subthalamic nucleus and globus pallidus.24,80,81 These phenotypes, while milder than those from overexpressed mHTT fragments, are comparable to HD knockin mouse models, 82 supporting that loss of normal HTT function contributes to HD pathogenesis. Even allele-selective strategies, therefore, require rigorous evaluation of their long-term effects on homeostatic processes, which remain still poorly characterized. This underscores the critical need for future studies to employ model systems in which the mutant allele is endogenously expressed and replaces one wild-type allele, thereby faithfully recapitulating the genetic context of HD patients. Therefore, while HTT-lowering remains viable, prospective studies must define dose-response relationships, cell-type-specific vulnerabilities, and long-term functional outcomes in vivo before clinical translation. The optimal therapeutic may not be maximal HTT suppression, but rather a finely titrated reduction that preserves essential HTT functions while mitigating mHTT toxicity.
Overall, an understanding of the interaction between HTT levels and mHTT toxicity across developmental stages through adulthood will be crucial for the definition of therapeutic windows for the restoration of wild-type HTT function or for increasing compensatory capacity.
Footnotes
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Hereditary Disease Foundation Research Grant (HDF 990846, MC); Fondation pour la Recherche Médicale (FRM: équipe labellisée DEQ202203014675, SH) ; Agence Nationale pour la Recherche (CortexDEVHD: ANR-23-CE16-0014-01, SH).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.