
Abstract
Huntington's disease (HD) is a neurodegenerative disorder triggered by an unstable expansion of CAG repeats in the coding sequence of the HTT gene. Among the neuronal populations affected, striatal spiny projection neurons (SPNs) show particular susceptibility to the pathogenic mutation. The basis of this selective vulnerability, however, is still not fully understood. In this review, we highlight recent epigenomic research on HD, framing it within current concepts in epigenetics. We propose that epigenetic regulation contributes to neuronal vulnerability in HD. Functional genomics studies provide growing evidence in support of this view. Findings from both HD animal models and patient-derived tissues have shown somatic CAG expansion-dependent epigenetic and transcription erosion in vulnerable neurons, which leads to accelerated loss of the identity of vulnerable neurons, promoting their premature aging. Moreover, HD stem cell models show that epigenetic and transcriptional alterations also occur during neural differentiation, impacting neuronal specification and maturation trajectories. This suggests that abnormal epigenetic priming during neurodevelopment might further predispose vulnerable neurons to later epigenetic and transcriptional erosion, thereby amplifying their susceptibility in the disease.
Keywords
Epigenetic regulation: general rules
Epigenetics refers to heritable modifications in gene expression that occur without altering DNA sequence. These processes shape transcription, replication, and repair through structural regulation of chromatin, a complex of DNA, RNA, and histone proteins. Histone modifications and DNA methylation play central roles in this regulation. 1 Chromatin is organized into nucleosomes, each composed of histone octamers (H2A, H2B, H3, H4). Histones undergo diverse post-translational modifications (PTMs), including acetylation, methylation, phosphorylation, and ubiquitination. These marks alter nucleosome compaction, thereby controlling chromatin accessibility to transcriptional machinery. The reversible and site-specific nature of these modifications, often interpreted by specialized “reader” proteins, forms the basis of the “histone code”.2,3 For instance, acetylated histone marks such as H3K27ac and methylation at H3K4 are linked to open, transcriptionally active chromatin (euchromatin), whereas repressive marks like H3K9me3 and H3K27me3 are hallmarks of repressed chromatin (heterochromatin). Importantly, histone signatures can distinguish functional regulatory regions: for example, active promoters are enriched in H3K4me3, while active enhancers, distant regulatory regions to promoters, typically display H3K27ac and H3K4me1.
DNA methylation involves covalent addition of a methyl group to cytosines, particularly in CpG dinucleotides, which is catalyzed by DNA methyltransferases (DNMTs).4,5 DNMT1 maintains existing patterns, whereas DNMT3a and DNMT3b establish de novo methylation. Once considered irreversible, methylation is now recognized as dynamic: TET enzymes convert 5-methylcytosine into hydroxymethylcytosine (5-hmC), enabling active demethylation through DNA repair pathways. Notably, neurons display high levels of 5-hmC, reflecting the plasticity of their epigenetic landscapes.6,7 Methylated CpG regions recruit methyl-binding proteins such as MeCP2, which interact with co-repressor complexes, including HDACs, to enforce gene silencing. CpG islands in promoter regions are particularly sensitive to this regulation and contribute to stable transcriptional repression.
Moreover, the chromatin is packaged within the 3D space of nuclei and subject to define hierarchical organization, ranging from broad compartments to smaller domains called topologically associating domains (TADs).8,9 TADs are themselves subdivided into subTADs, including chromatin loops that physically bring together promoter and enhancers to boost transcriptional activity. In vertebrates, CTCF plays a critical role in the establishment of 3D chromatin organization, in cooperation with the cohesin complex. 10
Technological advances over the past two decades have enabled genome-wide mapping of epigenetic features, including histone modifications and chromatin modifiers (e.g., by using ChIP-seq, CUT&Tag, CUT&RUN), chromatin accessibility (e.g., by using ATAC-seq), DNA methylation (e.g., by using BS-seq), and higher-order chromatin architecture (e.g., by using Hi-C). However, applying these approaches at cell population or single-cell resolution and integrating multi-omics data remain challenging. Yet, continued progress in single-cell technologies and bioinformatics promises to refine epigenomic profiling in a short term.11–13
Epigenetic regulation drives cellular identity acquisition
The establishment of cell type–specific identities during development relies on epigenetic regulation.14,15 This process is particularly intricate in the mammalian brain, where diverse neuronal subtypes emerge across specialized regions. Differentiation of neural stem cells involves dynamic transcriptional reprogramming orchestrated by temporally regulated transcription factors that build gene regulatory networks and drive transitions toward mature neuronal states.14,16 Epigenetic mechanisms coordinate these events by modulating chromatin accessibility in response to both intrinsic and extrinsic cues. Multiple layers of regulation—including histone modifications, DNA methylation, and higher-order chromatin organization—interact to ensure precise control of gene expression.17,18 Together, these principles underpin the acquisition and maintenance of stable cellular identity.
More specifically, the active enhancer marks H3K4me1 and H3K27ac are critical for establishing and maintaining cell identity. 17 During differentiation, the histone methyltransferases MLL3/MLL4 and histone acetyltransferases CBP/P300 deposit these marks at cell-type-specific enhancers, including super-enhancers, which are broad enhancers regulating cellular identity genes, as well as activity-regulated enhancers, responding to environmental stimuli.19,20 In the brain, activity-regulated genes, including immediate early genes (IEGs) and late responsive genes (LRGs), drive neuronal plasticity and functions like learning and memory.21,22 Pioneer transcription factors guide H3K4me1 and H3K27ac deposition at cell-type-specific enhancers during cellular differentiation, by binding closed chromatin and recruiting chromatin remodelers and histone modifiers.23–25 The promoter mark H3K4me3 is less dynamic than enhancer marks during differentiation. 17
Repressive marks also shape cell identity. H3K27me3, which is deposited by the polycomb repressive complex 2 (PRC2), silences developmental genes and co-occurs with H3K4me3 at bivalent promoters, providing epigenetic plasticity for cell lineage specification and maturation, including neural maturation.16,26,27 Ubiquitination of H2AK119 (H2AK119ub), a modification mediated by the polycomb repressive complex 1 (PRC1), and H3K9me3 promote chromatin compaction, transcriptional repression and genome integrity, supporting lineage specification and heterochromatin maintenance.28,29 DNA methylation is critical for silencing and cell fate restriction. TET-mediated oxidation produces 5hmC at active genes, particularly in post-mitotic neurons, shaping enhancer activity and neuronal diversity together with MeCP230–32
While previously thought stable, increasing studies show that TADs also reorganize during development, highlighting dynamic spatial regulation of gene expression.8,18,33
Crosstalk among histone modifications, DNA methylation, and chromatin architecture ensures proper acquisition and maintenance of cell identity.16,34
Loss of epigenetic information causes cellular aging
Cellular aging involves multiple interconnected hallmarks, including loss of stemness, senescence, inflammation, metabolic dysfunction, proteostasis decline, loss of genomic integrity, including telomere attrition and epigenetic alterations such as changes in DNA methylation, histone modifications and disruption of 3D chromatin.35–38 Although causal mechanisms of cellular aging remain debated, it is accepted that loss of genomic integrity is central. The somatic mutation theory attributes aging to cumulative DNA lesions, whereas the “information theory of aging” proposes that progressive loss of epigenetic information underlies cellular identity decline.38–40
Evidence from genetically engineered mice (i.e., inducible change to the epigenome or ICE mice) supports this latter view, showing that DNA break accumulation can induce aging-associated epigenetic erosion across DNA methylation, histone modification, and 3D chromatin architecture layers, without causing mutations. 41 Specifically, ICE mice display accelerated DNA methylation clock, as well as reduced super-enhancer activity due to depleted H3K27ac and weakened promoter–enhancer contacts and diminished H3K27me3 at developmental gene promoters, leading to identity gene repression and developmental gene reactivation, respectively. Such alterations are conserved across tissues, including brain. Additional studies support these results, showing that aging cells undergo de-repression of developmental genes enriched in PRC2-bound chromatin regions, including bivalent domains, driven by alterations in H3K27me3 and DNA methylation.42,43
Remarkably, suppression of H3K27me3 in striatal neurons, through inactivation of PRC2 de-represses developmental genes and downregulates striatal identity genes, suggesting that loss of PRC2 is sufficient to accelerate aging-related epigenetic erosion in striatal neurons. 44 In contrast, CBP/P300 loss decreases H3K27ac at neuronal super-enhancers without inducing developmental gene reactivation, 45 suggesting PRC2 and CBP play distinct yet complementary roles in cellular identity maintenance. Aging also blunts neuronal activity-dependent gene regulation in CBP-dependent manner. CBP depletion reduces H3K27ac at plasticity enhancers, impairing IEG and LRG induction and memory, particularly in aged neurons. 45 Dysregulation of additional histone acetylation marks (e.g., H4K12ac, H3K9ac) contributes to cognitive decline,46–49 highlighting loss of histone acetylation at plasticity genes as a hallmark of neuronal aging.
Constitutive heterochromatin, which is rich in repetitive elements, also deteriorates with age, losing nuclear lamina interactions and silencing capacity, resulting in retrotransposon activation.38,50,51 The mechanism involves reduction of H3K9me3, H3K27me3, and DNA methylation, depending on repeat type.
The link between DNA damage and epigenetic erosion remains elusive, but may involve sirtuins, NAD+-dependent deacetylases that are critical in maintaining genomic stability and chromatin organization. 52 It is thought that age-associated NAD+ decline and sirtuin dysfunction might drive loss of epigenetic information and cellular aging. 53
Epigenetic and transcriptional erosion in HD vulnerable neurons
Transcriptional erosion in HD vulnerable neurons
Transcriptomic analysis on allelic series of HD knockin (KI) mice shows that transcriptional dysregulation is progressive, CAG length-dependent, and most pronounced in the striatum, correlating with HD pathogenesis (Langfelder et al., 2016). Cell-type-specific and single cell approaches on HD vulnerable tissues, including post-mortem brain tissues of HD patients, reveals that striatal spiny projection neurons (SPNs) exhibit profound transcriptional dysregulation, compared to other cell types, and dysregulated genes display signature reflecting aging-related transcriptional erosion that promotes cell identity loss.54–58 Notably, downregulated genes are enriched in SPNs identity genes, while upregulated genes include developmental and senescence genes. Dysregulated genes in HD SPNs display additional signatures related to neuronal innate immunity, energy metabolism, DNA repair, autophagy and stress response, consistent with aging-related mechanism.55,57 Transcriptional dysregulation in glial cells is less extensive than in vulnerable neurons.54,55,57 Yet, HD astrocytes elicit protective transcriptional response, while oligodendrocyte identity marker genes are found down-regulated in HD oligodendrocytes.59–61
Transcriptional erosion in SPNs is linked to somatic CAG instability. The causing mutation in HD, a CAG expansion, undergoes cell-type-specific somatic expansion, which is most pronounced in vulnerable neurons such as SPNs, likely due to intrinsic features of these cell types, including elevated levels of genes driving somatic expansion (e.g., mismatch repair genes such as MSH2 and MSH3). Somatic CAG expansion in HD vulnerable neurons leads to continuous elongation of CAG expansion.57,62,63 Recent single-cell analyses using HD striatal tissue indicate that transcriptional erosion occurs once CAG repeats reach ∼150 units, with SPNs attaining this threshold faster than other striatal cells. 54 The study suggests that crossing the threshold triggers SPN identity loss and neurodegeneration, establishing somatic CAG expansion as a driver of HD. 54 In line with these results, Msh3 suppression in the HD KI mouse line Q140, expressing Htt with 140 CAGs, reduces both somatic CAG expansion and transcriptional erosion in SPNs, and mitigate disease progression, in contrast to inactivation of Msh3 in HD Q175 KI mice.58,64 However, recent data generated on HD patients indicates that down-regulation of SPN identity genes such as PDE10A and PCP4 occurs in most HD SPNs, suggesting lower CAG threshold drives transcriptional erosion, at least for some identity genes. 65
Epigenetic erosion in HD vulnerable neurons
Chromatin modifiers in HD
The hypothesis that epigenetic dysregulation contributes to HD emerged in the early 2000s, following the discovery that CBP interacts with HTT and is sequestered in polyQ-HTT nuclear aggregates in the brain of HD patients and mice.66,67 Preclinical studies further supported a role for histone acetylation in HD, showing beneficial effects of histone deacetylase (HDAC) inhibition and detrimental effects of inhibiting histone acetyl-lysine readers.68–72 Beyond CBP, polyQ-HTT modulates additional chromatin-modifying complexes, notably PRC2. HTT directly interacts with PRC2 and was found to enhance H3K27me3 deposition in a polyQ-length–dependent manner. 73 Dysregulation of other histone-modifying enzymes, including H3K9 methyltransferases, H3K4me3 demethylases and MeCP2, has also been reported,68,74–79 and transcriptomic data indicate multiple chromatin modifiers are altered in HD vulnerable neurons.80,81 Proteomic analyses further show that HTT can interact with multiple partners, including transcription and chromatin factors, which might impact gene regulation via gain and/or loss of function mechanisms. 82
Among these factors, CBP and PRC2 appear to play critical role in HD. Meta-analyses show strong overlap between genes dysregulated in HD mouse brains and those altered in neurons lacking CBP or lacking PRC2 catalytic subunits Ezh1/Ezh2. 83 Moreover, HD SPNs and SPNs deficient for PRC2 both undergo aging-like transcriptional erosion, which is characterized by downregulation of SPN identity genes and de-repression of developmental/senescence genes.54,56,80,84,85 Finally, single-nucleus RNA-seq on striatal tissues from HD patients and mice shows that dysregulated gene promoters are enriched for PRC2 targets, including bivalent promoters.55,56,58 These findings support a role for CBP and PRC2 dysfunction in transcriptional erosion of HD vulnerable cells.
Histone modifications and chromatin accessibility in HD
Epigenomic analyses are consistent with this hypothesis. ChIP-seq data generated using bulk striatal tissue of HD mice shows depletion of H3K27ac, a bona-fide target of CBP, at enhancers of SPN identity gene (i.e., super-enhancers).84,86 This depletion correlates with reduced RNA polymerase II recruitment and transcriptional downregulation of SPN identity genes. Additionally, H3K4me1 and H3K4me3 are reduced at enhancers and promoters of SPN identity genes, respectively.78,84 Moreover, 3D chromatin architecture analysis using 4C-seq shows disrupted spatial organization at some SPN identity genes, including Pde10a, due to weakened promoter–enhancer interactions, potentially linked to altered enhancer RNA regulation.86,87 Furthermore, temporal profiling of H3K27ac and PRC2-mediated H3K27me3, performed using ChIP-seq or CUT&Tag coupled with fluorescence-activated nuclear sorting (FANS), provide evidence for accelerated epigenetic erosion underlying transcriptional erosion in SPNs of Q140 mice starting as early as prodromal stage (e.g., 2 months), suggesting epigenetic changes precede behavioral alterations.86,88 More specifically, the epigenetic age, estimated using a composite score derived from H3K27ac and H3K27me3 signals at de-repressed developmental gene loci, is accelerated by approximately three months in 6-month-old Q140 SPNs compared to controls. 88 The data suggest a subset of bivalent developmental transcription factors initiates de-repression of developmental genes, through a mechanism involving PRC1. 88 Single nuclear ATAC-seq indicates epigenetic erosion in HD SPNs is causally linked to somatic CAG expansion, since transcriptionally active chromatin at identity genes and repressive chromatin at developmental genes are restored in Msh3-deficient SPNs of HD Q140 mice. 58
Genome-wide analysis of histone modifications using human HD brain remains limited. Nonetheless, post-mortem striatal tissue of Vonsattel grade 2 HD patients exhibits reduced H3K27ac at downregulated neuronal genes, 89 while increased H3K4me3 at bivalent developmental genes was reported, 90 consistent with mouse findings. Moreover, ATAC-seq data generated on FANS sorted brain cells from HD patients confirms loss of chromatin accessibility at H3K27ac-enriched cell-type-specific enhancers, including super-enhancers, in neurons that undergo somatic CAG expansion such as SPNs. 65 The study further indicates that gain of DNA methylation is involved in transcriptional repression of super-enhancer-regulated genes in HD SPNs (see also DNA methylation section below). Together, these data support the view that somatic CAG expansion in neurons of both HD mice and HD patients drives accelerated epigenetic and transcriptional erosion.
Cellular aging impairs adaptive gene regulation, including stress response and energy metabolism homeostasis, and, in the brain, neural plasticity. In HD mouse striatal neurons, transcriptional regulation of such genes progressively deteriorates.55,57,80,88,91 At epigenetic level, disruption of 3D chromatin architecture and dysregulation of H3K9ac at activity-regulated genes involved in neural plasticity is observed in HD mice, which correlates with behavioral deficits, particularly altered striatum-dependent memory processing. 92 These observations suggest an interplay between epigenetic/transcriptional erosion and dysregulation of adaptive gene programs in HD vulnerable neurons.
DNA methylation in HD
DNA methylation analyses in HD animal models and patient tissues indicate widespread alterations in methylation patterns and an accelerated epigenetic aging trajectory.65,93–97 Early studies examining a limited set of CpG sites reported accelerated epigenetic clock in cortical regions of HD patients, with multivariate modeling estimating an increase in epigenetic age of approximately three years attributable to HD status. This acceleration was observed in Vonsattel grade 1 tissues, indicating the mechanism is early. In contrast, results from striatal tissue were inconclusive, possibly reflecting extensive neuronal loss. 94 More recently, large-scale methylome datasets generated by reduced representation bisulfite sequencing (RRBS) and custom methylation arrays have expanded our understanding of DNA methylation changes in HD. 95 Notably, assessment of human blood methylomes using reference epigenetic clocks revealed accelerated epigenetic aging in manifest HD patients, suggesting that this component of accelerated aging is at least partly independent of somatic CAG repeat instability, which is minimal in blood cells. An epigenome-wide association study (EWAS) performed on HD blood samples identified 33 CpG sites predictive of HD mutation status. The most significant site, validated across various species and DNA sources, was located within exon 1 of HTT, adjacent to the CAG expansion. Additional differentially methylated CpG sites mapped to genes implicated in proteostasis and modulation of polyQ–HTT aggregation. Moreover, EWAS of motor progression in manifest HD revealed significant associations between disease progression and methylation levels at CpG sites near PEX14, GRIK4, and COX4I2. 95 Collectively, these findings demonstrate that both aging-associated and HD-specific CpG sites exhibit altered methylation patterns in HD tissues.
Moreover, recent data linked impairment in DNA methylation to transcriptional dysregulation in HD. Specifically, multi-omics analysis on FANS sorted striatal and cortical cells of HD patients indicate that DNA methylation changes contribute to neuronal identity erosion. 65 Oxidative bisulfite sequencing (oxBS-seq) was used to quantitatively assess cytosine methylation and hydroxymethylation at single nucleotide resolution. The data show that methylation changes are greatest in HD SNPs, when compared to non-affected cells such as glial cells, and that these changes correlate with chromatin accessibility changes. More precisely, super-enhancer-regulated genes such as PDE10a and PCP4 undergo gain of DNA methylation in HD SPNs, which associates with reduced ATAC signal and decreased transcription. Intriguingly, quantification of the effect indicates that most HD SPNs show altered DNA methylation profile at super-enhancer-regulated genes, suggesting the mechanism does not require long CAGs (e.g., >150 repeat units). Finally, the study suggests loss of MeCP2 binding is involved in transcriptional de-repression in HD SPNs. 65
Epigenetic and transcriptional regulation during neural differentiation in HD
Accumulating evidence indicates that abnormalities during HD brain development contribute to later onset of the neurodegenerative phenotype. 98 Specifically, human HD embryos display cortical defects, including disorganized germinal zones, impaired progenitor polarity, and mutant HTT accumulation in abnormal subcellular compartments. 99 This corroborates analyses showing that brain malformations are more frequently observed in HD patients than control individuals. 100 Moreover, preventing transient developmental defects induced by the HD gene in mice, enhancing neural circuit using ampakine or suppressing mutant HTT during developmental window is sufficient to delay the pathogenesis in the adult.101,102 Whether epigenetic underpinning underlies developmental anomalies in HD remains unclear. However, stem cell-derived neural cells and cerebral organoids provide some insight into this question.
Neuronal fate acquisition is impaired in HD iPSC-derived cortical and striatal neurons, and this associates with altered epigenetic and transcriptional developmental programs.103–109 Bulk RNA-seq and ChIP-seq analyses of HD iPSC-derived neurons expressing striatal neuronal markers reveal CAG-length-dependent altered neuronal maturation, resulting from dysregulation of developmental and synaptic genes. 105 Dysregulated genes display specific H3K4me3 and H3K27ac signatures – characterized by broad profiles – suggesting lineage-specific genes (super-enhancer-regulated genes) are more specifically impaired. 105 Further, multi-omics analysis, including snRNA-seq, shows persistent mitotically active population in HD iPSCs-derived cell cultures enriched in striatal neurons. 107 Interestingly, this population, expressing cyclin D1 and presenting elevated H3K4me3 and transcriptional levels at genes linked to cell cycle, stemness or development, arises in cells expressing HTT with CAG repeat expansion <150 (e.g., 46 and 53 repeats).
Further, bulk RNA-seq analysis on HD telencephalic organoids generated from the isogenic hESC RUES2 cell series, including control lines and three HD lines (with 48, 56 and 72 CAGs), shows immature transcriptional signature, characterized by impaired down-regulation of pluripotency markers and reduced induction of transcriptional regulators of neuronal differentiation. 103 More recently, single cell transcriptomics has revealed neurodevelopmental abnormalities in the ventral fate acquisition of HD telencephalic organoids resulting in fewer gabaergic neurons, and milder alteration affecting maturation of dorsal populations. 108 These data might suggest abnormal epigenetic trajectory during neural differentiation of HD organoids, particularly impacting the identity and function of ventral populations from which the striatum originates. Finally, multi-omic analysis, including transcriptomics, proteomic and metabolomics using human HD iPSC-derived cerebral organoids, reveals specific molecular signatures associate with neurodevelopmental defects. 109 More specifically, the data indicate that mutant HTT causes hypermetabolism upon initiation of neural commitment, likely due to dysregulation of genes such as CHCHD2, involved in mitochondrial integrated stress response. This suggests energy metabolism might early contribute to epigenetic and transcriptional mechanism involved in neurodevelopmental perturbation in HD.
Alternatively, epigenetic mechanism might directly contribute to HD neurodevelopmental defect, especially since HTT modulates PRC2 activity.73,110 HTT is required to shape bivalency, i.e., H3K27me3 and H3K4me3 mark deposition at developmental gene promoters during neural differentiation, with mutant HTT impairing this mechanism in polyQ-dependent manner. 111 Dynamic regulation of bivalency enables waves of activation/repression of developmental transcription factors, which promotes cellular state transitions during differentiation. Proper timing of this mechanism is especially critical during neuronal differentiation, where epigenetic barrier regulating bivalent genes determines neuronal maturation timeline, notably setting protracted pace, a feature of neuronal maturation. 112 Thus, mutant HTT might impair this timing. To disentangle loss- vs gain-of-HTT function, live cell imaging was performed together with multi-omics on cortical neurons (eCNS) derived from the isogenic hESC RUES2 cell series, which enabled to correlate phenotypic alterations with epigenomic, transcriptomic and proteomic changes induced by loss of HTT vs CAG expansion (ranging 56–72) during neural differentiation. 113 ATAC-seq and H3K4me1, H3K4me3, H3K27ac and H3K27me3 ChIP-seq data show a greater number of differential peaks for CAG expansion comparison vs controls, than for HTT KO comparison vs controls. Nonetheless, convergent biological processes, especially linked to development, are enriched in both comparisons. Integrative analysis using network-based approach further supports similarities between biological processes dysregulated in CAG-expanded and HTT KO eCNs, including neuronal differentiation, cell fate commitment and DNA replication, implicating developmental transcription factors such as SP1 and LHX3. Network analysis also reveals pathways that are specific to each genotype, linked to axon guidance in HTT KO, and metabolic processes in CAG expansion condition. 113 Finally, DNA methylation analyses have revealed altered methylation profiles during neuronal differentiation of HD cells, pointing to hypermethylation of chromatin modifiers involved in differentiation, including genes related to H3K27 and H3K4 demethylation or methylation.96,112,114 Together, this suggests coordinated epigenetic and transcriptional response is involved in abnormal neurodevelopmental features of HD neurons, through a combination of gain- and loss-of-HTT function that may affect timeline of neural differentiation, due to impaired dynamic regulation of bivalent promoters.
Conclusions and perspectives
Omic approaches have substantially advanced our understanding of the epigenetic landscape in HD cells. An aging-associated epigenetic and transcriptional erosion signature is evident in both HD patients and mouse models (Figure 1). This erosion, which establishes in neurons that undergo somatic CAG expansion, leads to a loss of neuronal identity and drives neurodegeneration. Moreover, transcriptional and epigenetic alterations are observed during neural differentiation of HD stem cells, which might underlie neurodevelopmental component to HD (Figure 1). However, many questions remain unresolved. Precise mechanisms driving epigenetic and transcriptional dysregulation during neural differentiation and neuronal aging in HD is unclear. The role of HTT in these mechanisms needs to be specified. Mutant HTT may affect cell state transitions during neural differentiation, impairing bivalency regulation. Alternatively, and not exclusively, it might impact energy metabolism, including mitochondrial function, which is critical upon initiation of neural commitment. The mechanism underlying aging-related epigenetic and transcriptional erosion in HD vulnerable neurons is also elusive, though evidence indicates that CBP and PRC2 are implicated. Recent studies show that the mechanism requires somatic CAG expansion, but it remains debated whether specific CAG threshold needs to be reached, and whether polyQ-HTT aggregates are required. Last, whether developmental and aging-related mechanisms independently or synergistically modulate neuronal vulnerability in HD is currently unknown. It is plausible that early perturbations, even subtle, during neural cell lineage specification and maturation further sensitize vulnerable neurons to later somatic CAG expansion-dependent loss of epigenetic information, resulting in premature aging. It is tempting to speculate that anomalies during differentiation of HD neural cells induce long-term epigenetic priming at genes involved in adaptive stress response, thereby contributing to accelerated aging of HD vulnerable neurons. To address these questions, it will be critical to perform comprehensive, high resolution multi-omics analyses, through time series at single-cell or cell population levels, including early/developmental stages. Given that multiple epigenetic layers—including histone modifications, DNA methylation and 3D chromatin organization—coordinate gene regulation, elucidating how they interplay in HD cells will be key to uncover mechanisms. Human HD stem cell-derived models, including organoids, as well as mouse models proved valuable model systems and will likely remain critical to investigate early, including developmental, stages, given the difficulty to access human brains.
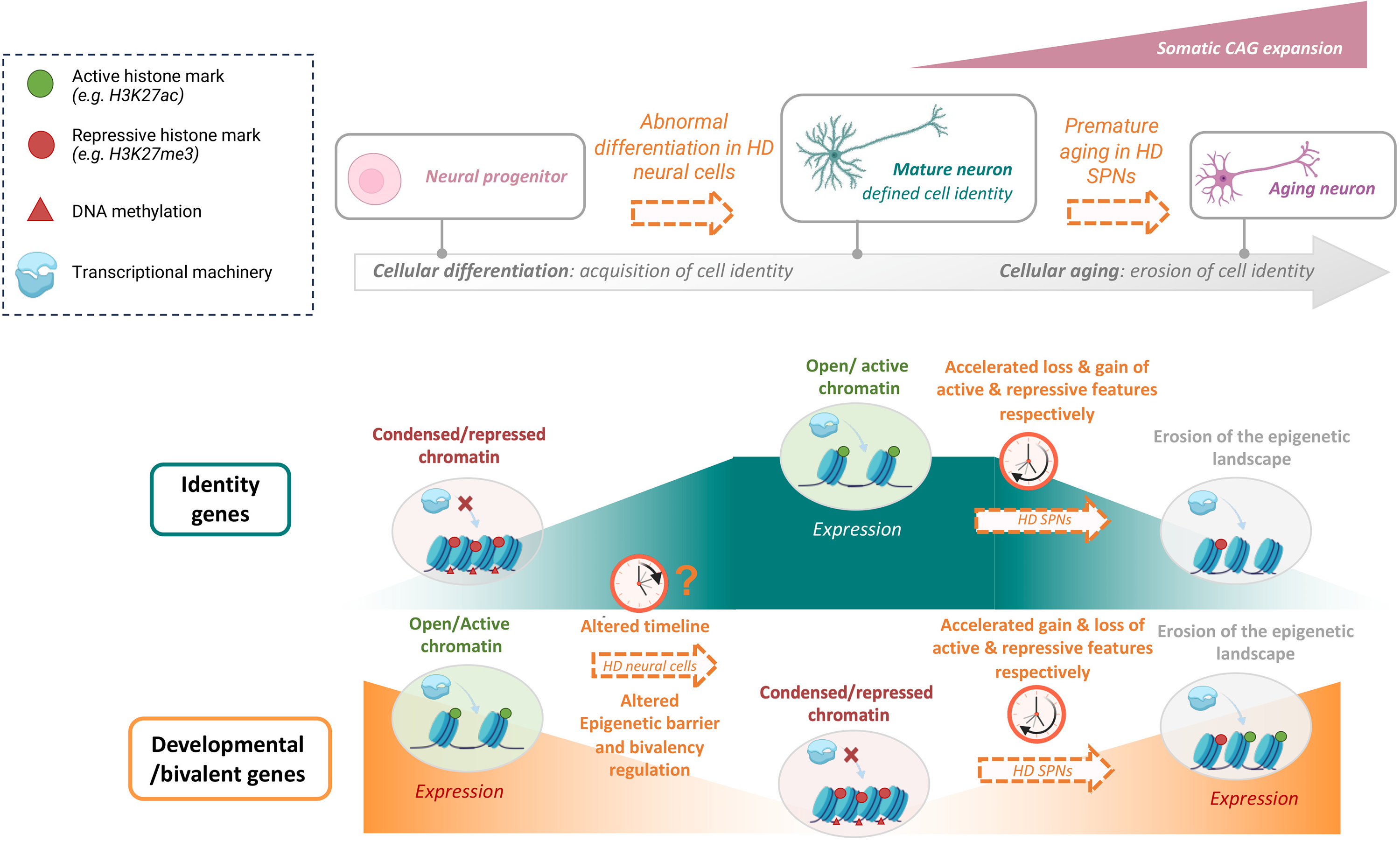
Epigenetic and transcriptional dysregulation in hd vulnerable neurons.
Schematic representation of chromatin states during neuronal differentiation and aging. The HD gene leads to neurodevelopmental anomalies, which might involve altered epigenetic barrier due to dysregulation of gene bivalency, leading to impaired pace of neural cell differentiation. Somatic CAG expansion in HD vulnerable neurons (e.g., SPNs) leads to premature aging, as a result of accelerated epigenetic and transcriptional erosion. HD SPNs display accelerated loss and gain of active and repressive chromatin features at identity genes, and conversely, gain and loss of active and repressive chromatin features at developmental genes.
Footnotes
Acknowledgements
N.P. and J.S. are recipients of doctoral fellowships from the agence nationale de la recherche (ANR) and French government, respectively. K.M. research is supported by the Agence Nationale de la Recherche [ANR-2022-CE12-0033], the Fondation de la Recherche Medicale (FRM), the Interdisciplinary Thematic Institute NeuroStra, the Centre National de la Recherche Scientifique (CNRS) and the university of Strasbourg.
Ethical considerations
N.A.
Consent to participate
N.A.
Consent for publication
N.A.
Funding
N.P. and J.S. are recipients of doctoral fellowships from the agence nationale de la recherche (ANR) and French government, respectively. K.M. research is supported by the Agence Nationale de la Recherche [ANR-2022-CE12-0033]; the Fondation de la Recherche Medicale (FRM); the Interdisciplinary Thematic Institute NeuroStra.
Agence Nationale de la Recherche, Fondation pour la Recherche Médicale, ITI NeuroStra, Strasbourg university, (grant number ANR-2022-CE12-0033).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
N.A.