
Abstract
Background
Acute lung injury (ALI) is a severe clinical syndrome characterized by excessive inflammation and high mortality. The TLR4/MyD88/NF-κB signaling pathway plays a pivotal role in the pathogenesis of LPS-induced ALI. This study aimed to investigate the protective effects of apigenin (API) against LPS-induced ALI in mice and to explore whether these effects are mediated through the inhibition of the TLR4/MyD88/NF-κB pathway.
Methods
An ALI model was established in C57BL/6 mice via intratracheal instillation of LPS (10 mg/kg). Mice were pretreated with API at doses of 25, 50, and 100 mg/kg or dexamethasone (5 mg/kg) via intraperitoneal injection. Assessments included lung wet/dry weight ratio, bronchoalveolar lavage fluid (BALF) protein concentration, Evans Blue leakage assay, histopathological evaluation, ELISA for inflammatory cytokines TNF-α, IL-6 and IL-1β, as well as oxidative stress markers MDA, SOD, CAT, GSH-Px, and qPCR analysis of TLR4, Myd88, and NF-κB mRNA expression, and Western blot for TLR4, MyD88, total NF-κB p65, and phosphorylated NF-κB p65 (p-NF-κB p65) protein levels.
Results
API pretreatment dose-dependently attenuated LPS-induced pulmonary edema, vascular hyperpermeability, and histopathological damage. API significantly suppressed the elevated levels of pro-inflammatory cytokines TNF-α, IL-6 and IL-1β in BALF and mitigated oxidative stress by reducing MDA content while enhancing the activities of antioxidant enzymes SOD, CAT and GSH-Px. Furthermore, qPCR analysis revealed that API inhibited the LPS-induced upregulation of Tlr4, Myd88, and Nfkb1 mRNA expression in lung tissue. Western blot analysis confirmed that API dose-dependently inhibited LPS-induced upregulation of TLR4, MyD88, and p-NF-κB p65 protein levels.
Conclusion
Apigenin effectively attenuated LPS-induced acute lung injury in mice by inhibiting the TLR4/MyD88/NF-κB signaling pathway, thereby reducing inflammation and oxidative stress. These findings provide preclinical evidence supporting the potential of Apigenin as a therapeutic agent for ALI.

1. Introduction
Acute lung injury (ALI) is a clinical syndrome characterized by inflammatory responses and high mortality. Its complex molecular and pathological mechanisms involve multiple signaling pathways.1-3 Studies have shown that cGAS-STING participates in regulating Neutrophil Extracellular Traps-mediated inflammatory lung injury, 4 and exosome-derived Tenascin-C triggers macrophage pyroptosis, amplifying inflammation during sepsis-induced acute lung injury. 5 Recent studies have further elucidated the role of programmed cell death pathways in ALI, including apoptosis, pyroptosis, necroptosis, and PANoptosis, which contributes to the pathogenesis of both bacterial and viral induced lung injury,6,7 such as influenza A virus dissemination can lead to tissue resident cell injury through activation of the PANoptosis pathway. 8 Conversely, some mechanisms offer protection. The gut microbiota can play a protective role in lipopolysaccharide (LPS)-induced ALI by regulating the TLR4/NF-κB signaling pathway. 9 Furthermore, targeting CSE1L can inhibit SP1 transcriptional activity, thereby alleviating macrophage inflammation. 10 In vascular endothelial cells, PPARδ can reduce vascular leakage by inhibiting CXCL10, 11 and S-glutathionylation of FABP5 promotes its interaction with PPARβ/δ, activates PPARβ/δ target genes, and suppresses lipopolysaccharide-induced macrophage inflammation, mitigating ALI. 12 Additionally, interfering with the expression of either Srg3 or Metrnβ can affect the progression of ALI by regulating ferroptosis and activating the SIRT1-p53 pathway, respectively.13,14
As research has progressed, various compounds and their targets have been confirmed to have ameliorative effects on ALI. Studies have shown that bacterial proteases and chymotrypsin can also alleviate oxidative stress, edema, and fibrosis by modulating the TLR4/Nrf2/NF-κB signaling pathway, thereby alleviate acute lung injury. 15 Celastrol treatment also significantly alleviates pulmonary tissue inflammation and improves sepsis-induced ALI by inhibiting the NF-κB/HIF-1α pathway, lowering IL-1β and TNF-α expression. 16 Andrographolide exerts anti-inflammatory and anti-ferroptosis effects in LPS-induced ALI by targeting TLR4 and regulating the Keap1/Nrf2 pathway. 17 Notably, multiple studies have confirmed that the TLR4-mediated inflammatory signaling pathway plays a crucial role in the inflammatory cascade of ALI. Inhibition of the S100A9 gene suppresses macrophage M1 polarization and sepsis via the TLR4/MyD88/NF-κB pathway, thereby alleviating LPS-induced ALI. 18 Trelagliptin, as well as novel nano-drugs Ber-lipo and Pac-lipo can alleviate inflammation and oxidative stress by modulating the TLR4/NF-κB signaling pathway, thereby mitigating ALI.2,19,20
Among numerous substances with therapeutic potential, natural flavonoids have attracted considerable attention due to their pronounced anti-inflammatory and antioxidant activities. Studies indicate that various flavonoid components exhibit protective effects in lung injury. For instance, procyanidin B2 alleviates lung Ischemia/Reperfusion injury by modulating the SIRT3/PKM2 pathway, 21 Norwogonin alleviates inflammatory responses and restores vascular barrier function by inhibiting the Src/AKT1/NF-κB signalling pathway through targeting Src, AKT1, and COX-2, thereby improving LPS-induced acute lung injury. 22 Additionally, studies have shown that flavonoids such as quercetin, baicalin, and rutin can alleviate acute lung injury.23-25 Recent studies have further highlighted the therapeutic potential of various flavonoids in inflammatory diseases,26,27 reinforcing the interest in natural compounds for the treatment of ALI.
Apigenin (API) is a natural flavonoid compound widely distributed in various fruits and vegetables, which has been confirmed to exhibit potent antioxidant and anti-inflammatory activities. Research indicates that API may inhibit pyroptosis by modulating NLRP3 inflammasomes, 28 and alleviate retinal inflammation and allergic rhinitis via the TLR4/MyD88/NF-κB pathway.29,30 Moreover, accumulating evidence suggests that API exerts protective effects in multiple inflammatory conditions through modulation of key signaling pathways.31-33 However, its protective effects and precise molecular mechanisms in the LPS-induced ALI remain understudied. Therefore, this study aims to establish an LPS-induced mouse ALI model to systematically evaluate the intervention effects of different doses of API, and to investigate whether it exerts protective effects by regulating the TLR4/NF-κB signaling pathway.
2. Materials and Methods
2.1. Experimental Animals
SPF-grade C57BL/6 mice (6-8 weeks old, weighing 20-22 g) used in the experiment were purchased from Beijing Weishanglide Biotechnology Co., Ltd. (Beijing, China). All mice were housed in a clean environment with constant temperature (22 ± 2 °C) and humidity (55 ± 10%), housed under a 12-hour light-dark cycle. Before experiments, all mice received at least one week of acclimatization, with free access to standard rodent feed and water. All animal experimental procedures involved in this study were reviewed and approved by Zhinanzhen Biology Ethics Committee (Approval Number: A2024000336). The reporting of this study conformed to the ARRIVE 2.0 guidelines. 34
2.2. ALI Model Establishment and Grouping
The acute lung injury model was induced by intratracheal instillation of lipopolysaccharide (LPS). Mice were randomly divided into 6 groups using a random number table generated by SPSS software. The investigator performing the animal experiments, including LPS instillation, drug administration, and sample collection, and outcome assessments, was blinded to group allocation. A normal group was established, with mice receiving an intratracheal injection of an equal volume of sterile PBS. Other mice were anaesthetised with an intraperitoneal injection of sodium pentobarbital (30 mg/kg). LPS (10 mg/kg, 50 μL) dissolved in sterile phosphate-buffered saline (PBS) administered via tracheal injection.
35
Treatment groups received API (purity ≥98%, Sigma-Aldrich, USA) three times, respectively one day before, on the day of, and one day after LPS induction.
36
The doses of API (25, 50, and 100 mg/kg) were selected based on previous studies demonstrating its anti-inflammatory effects in murine models28,37 and preliminary dose-range finding experiments. The specific grouping is as follows (n=6): I Normal group: Intratracheal injection of PBS, intraperitoneal injection of vehicle (0.5% carboxymethyl cellulose sodium solution, CMC-Na). II LPS model group: Intratracheal injection of LPS, intraperitoneal injection of vehicle. III LPS + API low-dose group: Intratracheal injection of LPS, intraperitoneal injection of API (25 mg/kg). IV LPS + API medium-dose group: Intratracheal injection of LPS, intraperitoneal injection of API (50 mg/kg). V LPS + API high-dose group: Intratracheal injection of LPS, intraperitoneal injection of API (100 mg/kg). VI LPS + Dexamethasone (DXMS), positive control group: Intratracheal instillation of LPS, intraperitoneal injection of dexamethasone (5 mg/kg).
38
All mice were euthanized under deep anesthesia 48 hours after LPS treatment. Blood samples were collected from the retro-orbital venous plexus, centrifuged at 4 °C (3,000 × g, 15 minutes) to separate serum, and stored at -80 °C. Lungs were lavaged via tracheal intubation using 0.5 mL of ice-cold PBS, three times (total lavage volume 1.5 mL), to collect bronchoalveolar lavage fluid (BALF). BALF was centrifuged at 4 °C (1,000 × g, 10 minutes), and the supernatant was aliquoted for subsequent analysis. The right lung was isolated and weighed to calculate the wet/dry weight ratio. The left lung was divided into two parts: one part was fixed in 4% paraformaldehyde for histological analysis; the other part was rapidly immersed in liquid nitrogen for rapid freezing, then transferred to -80 °C storage for subsequent biochemical and molecular biological assays.
2.3. Assessment of Alveolar Fluid Clearance (AFC)
To evaluate alveolar epithelial fluid transport function and pulmonary edema, the AFC rate was measured. The mice were euthanized by cervical dislocation, and the trachea, lungs, and heart were carefully dissected. A tracheal cannula was inserted into the right lower lung lobe. An isosmolar 5% albumin solution (0.15 mg Evans Blue-labeled) in normal saline was instilled via the cannula at a dose of 4 mL/kg body weight. Subsequently, 2 mL of oxygen was administered to facilitate the distribution of the instillate into the alveolar spaces. The lung was then subjected to a ventilation process involving 100% oxygen, with the airway pressure maintained within the range of 650 to 700 Pa for a duration of 1 hour. Subsequent to the ventilation period, 0.2 mL of fluid from the right lower lobe was aspirated. The absorbance of this fluid was measured at 620 nm. The AFC rate was calculated using the following formula: AFC (%) = [(Instilled volume - Remaining alveolar volume)/Instilled volume] × 100%, where Remaining alveolar volume = Instilled volume × (Initial protein concentration/Final protein concentration).
2.4. Lung Wet/Dry Weight Ratio (W/D Ratio)
To quantitatively evaluate the degree of pulmonary edema, the wet/dry weight ratio of lung tissue was measured. Following excision of the right lung, the wet weight was immediately recorded. The samples were then placed in a 65 °C oven for 72 hours until constant weight was achieved, after which the dry weight was measured. The W/D ratio was calculated to evaluate the severity of pulmonary oedema.
2.5. Evans Blue (EB) Leakage Assay for Assessing Pulmonary Vascular Permeability
Equivalent volumes of BALF samples were mixed with twice the volume of formamide and incubated at 60 °C in a water bath for 24 hours, followed by centrifugation at 12,000 × g for 15 minutes. Similarly, serum samples were appropriately diluted with formamide and processed using the same experimental protocol. The supernatant was collected and the absorbance values were measured at 620 nm using a multifunctional microplate reader (BioTek, USA). The pulmonary vascular permeability index (BALF-EB/Serum-EB) was expressed as the ratio of two absorbance values.
2.6. Histopathological Analysis
Fixed lung tissues were embedded in paraffin, sectioned into 4-μm-thick sections, and stained with hematoxylin and eosin (H&E). Pathological alterations including alveolar wall thickening, hemorrhage, and inflammatory cell infiltration were observed under a light microscope. According to a previous study, a standardized lung injury scoring system was used for evaluation. 39 To ensure objectivity, six randomly selected microscopic fields per sample were analyzed using ImageJ software (NIH, USA) to quantify alveolar edema area ratio, alveolar collapse rate and inflammatory cell count, as well as the mean score was calculated. The alveolar edema area ratio (%) = (edematous alveolar area/total observed alveolar area) × 100; the alveolar collapse rate (%) = (number of collapsed alveoli/total number of counted alveoli) × 100. All histopathological evaluations were conducted by two independent pathologists blinded to the group allocation.
2.7. Enzyme-Linked Immunosorbent Assay (ELISA)
Quantitative measurement of tumour necrosis factor-α (TNF-α), interleukin-6 (IL-6) and interleukin-1β (IL-1β) concentrations in BALF supernatant were performed using a mouse ELISA kit (Jianglai, China). BALF supernatant samples and standards were added to microplates pre-coated with capture antibodies. After incubation and washing, biotin-labeled detection antibodies, horseradish peroxidase-labeled streptavidin, and substrate solution were sequentially added to initiate the colourimetric reaction.The absorbance of each well was measured at 450 nm using a microplate reader. Standard curves were plotted based on the concentration and absorbance values of the standards, and the concentrations of inflammatory cytokines in each sample were calculated accordingly.
To assess the level of oxidative stress in lung tissue, corresponding ELISA kits (Jianglai, China) were used to measure the content of malondialdehyde (MDA), superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GSH-Px) in lung tissue homogenates. MDA content was determined using the thiobarbituric acid (TBA) method. The contents of SOD, CAT, and GSH-Px were all detected using corresponding ELISA kits. All experimental procedures were performed in strict accordance with the manufacturer’s kit instructions. The lung tissue was weighed and homogenized in pre-chilled PBS at a 1:9 (w/v) ratio. The mixture was mechanically homogenised in an ice bath to produce a 10% tissue homogenate. After centrifugation, the supernatant was collected for subsequent analysis.
2.8. Real-Time Quantitative Polymerase Chain Reaction (qPCR) Analysis
The Primer Sequences of Mice
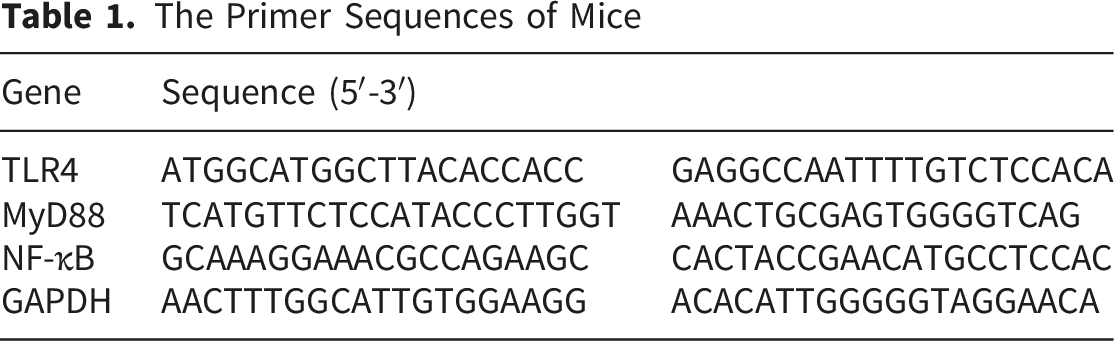
2.9. Western Blot Analysis
Lung tissue samples were lysed in RIPA lysis buffer (Beyotime, China) supplemented with protease and phosphatase inhibitors. The total protein concentration of the samples was determined using a BCA protein quantification kit (Solarbio, China). An aliquot of 30 μg of protein sample was separated by 10% SDS-PAGE and then transferred to a PVDF membrane (Millipore, USA). The membrane was blocked in TBST buffer containing 5% skim milk powder at room temperature for 1 hour, followed by overnight incubation with primary antibodies against TLR4, MyD88, NF-κB p65, phosphorylated NF-κB p65 (Ser536), and GAPDH (Cell Signaling Technology, USA). After washing with TBST for 3 times, the membrane was incubated with a secondary antibody conjugated to HRP at room temperature for 1 hour. Protein bands were visualized using an ECL reagent (Thermo Fisher Scientific, USA), and the bands were scanned and quantitatively analyzed using ImageJ software. The relative expression levels of the target proteins were normalized using GAPDH as an internal control. The activation level of the NF-κB signaling pathway was assessed by calculating the ratio of p-NF-κB p65 to total NF-κB p65. All samples were tested in triplicate.
2.10. Statistical Analysis
All data were expressed as mean ± standard deviation (mean ± SD). Statistical analysis was performed using GraphPad Prism software (version 10.1.2). Comparisons among multiple groups were conducted using one-way analysis of variance (ANOVA), with Tukey’s post hoc test for multiple comparisons. P < 0.05 was considered statistically significant.
3. Results
3.1. API Alleviates Pulmonary Edema and Vascular Leakage
Compared with the normal group (4.32 ± 0.40), the lung wet/dry weight ratio (W/D ratio) of mice with LPS-induced acute lung injury (6.10 ± 0.07) was significantly increased (P < 0.001). The AFC of normal group mice was 25.01 ± 1.04%, whereas the AFC of LPS group mice was significantly reduced to 10.85 ± 1.24% (P < 0.001). The pulmonary vascular permeability index of normal group mice was 0.26 ± 0.05, whereas the BALF-EB/serum-EB of LPS group mice was significantly increased to 1.92 ± 0.15 (P < 0.0001). These results indicated a significant increase in pulmonary edema and vascular permeability induced by LPS in mice. After API intervention, all dose groups (25, 50, and 100 mg/kg) exhibited a dose-dependent significant reduction in W/D ratio, AFC, and BALF-EB/serum-EB (P < 0.05). Among them, the high-dose API treatment group (100 mg/kg) showed the most significant effect, with W/D ratio, AFC, and BALF-EB/serum-EB being 4.34 ± 0.21, 19.38 ± 1.46%, and 0.93 ± 0.06, respectively, all significantly lower than those of the LPS model group (P< 0.0001) and comparable to those of the DXMS group. The results indicate that API can effectively alleviate LPS-induced pulmonary edema and vascular barrier damage (Figure 1). Apigenin attenuates LPS-induced pulmonary edema and vascular leakage in mice. (A) Lung wet/dry (W/D) weight ratio. (B) Alveolar fluid clearance (AFC) rate. (C) Pulmonary vascular permeability index assessed by the Evans Blue (EB) leakage assay. Data are presented as mean ± SD (n=6). * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001
3.2. API Ameliorates Pathological Damage in Lung Tissue
H&E staining revealed that, compared with the control group, LPS-induced model mice exhibited severe alveolar structural disruption, thickened alveolar walls, and extensive inflammatory cell infiltration in lung tissue (Figure 2A). API pretreatment significantly alleviated these pathological alterations. Semi-quantitative analysis of histopathological scores revealed that the LPS model group score was significantly higher than that of the normal group (P < 0.01). API pretreatment exerted a dose-dependent amelioration of these pathological alterations, with a clear dose-response improvement effect. Among them, the lung tissue morphology of the high-dose (100 mg/kg) API group was closest to normal, with effects comparable to the DXMS group (Figure 2B). Apigenin ameliorates LPS-induced histopathological changes in lung tissue. (A) Representative images of lung sections stained with hematoxylin and eosin (H, E) (scale bar = 100 μm). (B) Semiquantitative assessment of lung injury scores. (C) Quantitative analysis of lung injury indicators: the ratio of alveolar oedema area, the rate of alveolar collapse, and the inflammatory cell count. Data are presented as mean ± SD (n=3). * P < 0.05, *** P < 0.001
Treatment with the API improved these pathological changes in a dose-dependent manner. In the API group (25, 50, and 100 mg/kg), the ratio of alveolar oedema area, the rate of alveolar collapse and the inflammatory cell count were all significantly reduced compared with the LPS group (P < 0.001). The high-dose API group (100 mg/kg) demonstrated the most pronounced protective effect, comparable to those in the positive control group treated with DXMS (Figure 2C).
3.3. API Inhibits Inflammatory Responses
ELISA results demonstrated that LPS stimulation significantly increased levels of TNF-α (335.05 ± 28.20), IL-6 (73.13 ± 3.74), and IL-1β (188.06 ± 10.13) in BALF compared to normal group (TNF-α 63.49 ± 13.26, IL-6 13.21 ± 2.36, and IL-1β 17.45 ± 4.97). API pretreatment significantly reduced the levels of these pro-inflammatory cytokines in a dose-dependent manner. The highest dose group (100 mg/kg) exhibited the most pronounced inhibitory effect of TNF-α (101.25 ± 14.43), IL-6 (21.74 ± 3.47), and IL-1β (30.43 ± 2.19) in BALF compared to LPS group, P < 0.0001 (Figure 3). Apigenin reduces the levels of pro-inflammatory cytokines in BALF. Concentrations of (A) TNF-α, (B) IL-6, and (C) IL-1β in BALF were measured by ELISA. Data are presented as mean ± SD (n=3). **** P < 0.0001
3.4. API Inhibits Activation of the TLR4/MyD88/NF-κB Signalling Pathway
qPCR analysis revealed that LPS stimulation significantly upregulated the mRNA expression of TLR4 and MyD88 in lung tissues, and increased the expression of NF-κB. Compared to the control group, the expression of TLR4, MyD88, and NF-κB in LPS-treated lung tissues increased to 2.61, 2.78, and 2.77 fold, respectively. API intervention dose-dependently inhibited the mRNA expression of TLR4, MyD88, and NF-κB. In the 100 mg/kg API intervention group, the expression of TLR4, MyD88, and NF-κB decreased to 1.38, 1.21, and 1.09, respectively (Figure 4A). However, the regulatory effect of the DXMS group on the mRNA expression of TLR4, MyD88, and NF-κB was relatively weak. Apigenin inhibits the activation of the TLR4/MyD88/NF-κB signaling pathway in lung tissue. (A) Relative mRNA expression levels of TLR4, MyD88, and NF-κB were determined by qPCR. GAPDH was used as an internal control. (B) Representative Western blot bands and corresponding quantitative statistical analysis of TLR4, MyD88, NF-κB p65, and p-NF-κB p65. Data are presented as mean ± SD (n=3). * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001, ns, not significant
To confirm these findings at the protein level, we conducted Western blot analysis. As shown in Figure 4B, LPS challenge significantly increased the expression of TLR4, MyD88, and p-NF-κB p65 proteins, as well as the ratio of p-NF-κB p65 to total NF-κB p65. API treatment dose-dependently reduced these protein levels, with the high-dose group being closest to normal levels. The regulatory effect of DXMS on these proteins was weaker than that of high-dose API, which is consistent with the qPCR results.
3.5. API Inhibits Excessive Oxidative Stress Response
Compared with the control group (MDA 0.51 ± 0.08 ng/mL, SOD 5.83 ± 0.23 ng/mL, CAT 621.92 ± 37.89 pg/mL, GSH-Px 9.13 ± 0.52 ng/mL), MDA (2.06 ± 0.14 ng/mL) content increased, and SOD (2.19 ± 0.27 ng/mL), CAT (378.12 ± 11.12 pg/mL), and GSH-Px (3.31 ± 0.36 ng/mL) contents decreased in the lung tissue of the model group, P < 0.001. Compared with the model group, the API intervention groups exhibited significantly reduced MDA levels in lung tissue (P < 0.01), while SOD, CAT, and GSH-Px levels were significantly increased (P < 0.05), demonstrating a dose-dependent effect. Among them, the high-dose (100 mg/kg) API group showed the most significant recovery in oxidative stress levels (MDA 0.86 ± 0.06 ng/mL, SOD 4.87 ± 0.17 ng/mL, CAT 652.94 ± 54.4 pg/mL, GSH-Px 8.80 ± 0.40 ng/mL, P < 0.0001 vs. LPS group), with effects comparable to the DXMS group. The results indicate that API effectively alleviate LPS-induced oxidative stress damage (Figure 5). Apigenin mitigates LPS-induced oxidative stress in lung tissue. (A) Content of malondialdehyde (MDA). Activities of the antioxidant enzymes (B) superoxide dismutase (SOD), (C) catalase (CAT), and (D) glutathione peroxidase (GSH-Px). Data are presented as mean ± SD (n=3). * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001
4. Discussion
This study elucidates that API exerts protective effects in mitigating inflammation and oxidative stress via the TLR4/MyD88/NF-κB signalling pathway during LPS-induced ALI in mice. The experimental findings demonstrate that API can dose-dependently alleviate pulmonary edema, reduce vascular permeability, improve lung histopathological damage, inhibit the release of inflammatory cytokines, and mitigate oxidative stress responses.
The TLR4/MyD88/NF-κB pathway constitutes the core mechanism by which LPS triggers inflammatory responses.18,40 In this study, LPS significantly upregulated the mRNA expression of Tlr4, Myd88, and Nfkb1 in lung tissue, whereas API intervention significantly suppressed the expression of these genes. Western blot analysis confirmed that API also downregulated the protein levels of TLR4, MyD88, and phosphorylated NF-κB p65, as well as the ratio of p-p65 to total p65. This strongly demonstrates that API inhibits this pathway at transcriptional levels. This finding is consistent with previous studies reporting that API inhibits inflammation via the same pathway in arthritis and allergic rhinitis.30,41 The present study further confirms that TLR4/MyD88/NF-κB constitutes a pivotal target for anti-inflammatory effects of API. As a central transcription factor regulating inflammation, NF-κB activation promotes the expression of pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1β. 42 The observed reduction of TNF-α, IL-6 and IL-1β by approximately 15-30% in the high-dose API group is biologically significant, as they are important mediators of the acute phase response and endothelial activation in ALI. 43 This study showed that API significantly reduced the levels of these factors in BALF, functionally confirming API’s suppression of NF-κB downstream inflammatory responses. However, this study did not assess IκBα degradation or the nuclear translocation of p65, which are key events in NF-κB activation. This limitation should be addressed in future studies.
Furthermore, while our study focuses on the TLR4/MyD88/NF-κB pathway, we acknowledge that apigenin may exert its protective effects through alternative or additional pathways. For instance, apigenin has been reported to modulate the Nrf2/HO-1 pathway to exert antioxidant effects, 44 inhibit the NLRP3 inflammasome, 28 and regulate the JAK/STAT or MAPK signaling cascades.33,45 The potential contribution of these pathways to the observed anti-inflammatory and anti-oxidative effects in our ALI model cannot be ruled out and warrants further investigation.
In addition to inflammatory responses, oxidative stress constitutes a significant mechanism in the development of ALI. 46 LPS can induce the production of reactive oxygen species (ROS), leading to lipid peroxidation and impaired function of the antioxidant enzyme system. 47 In this study, API intervention significantly reduced MDA levels in lung tissue while enhancing the activity of SOD, CAT, and GSH-Px. This indicates that API not only mitigates oxidative damage but also bolsters endogenous antioxidant capacity. This effect may be related to the inhibition of the NF-κB pathway, as oxidative stress and inflammatory responses often form a positive feedback loop, synergistically exacerbating tissue injury. Moreover, recent studies have highlighted the involvement of the Nrf2/HO-1 pathway in mediating antioxidant effects of flavonoids.44,48 Although this study is not explored here, API may also activate the Nrf2 signaling pathway, thereby promoting the upregulation of antioxidant enzyme expression in lung tissue. Future research should explore this possibility.
This study elucidates that API exerts a lung-protective effect in an LPS-induced ALI by synergistically inhibiting both the TLR4/MyD88/NF-κB inflammatory pathway and oxidative stress pathways, confirming its dose-dependent efficacy. Compared to dexamethasone, API at a dose of 100 mg/kg demonstrated similar efficacy in reducing pulmonary edema, inflammation, and oxidative stress, indicating that API may be a potential alternative or adjuvant drug with fewer side effects. However, its duration of action and long-term effects still require further investigation. Furthermore, as a natural flavonoid compound, API offers advantages including broad availability and high safety profile. It shows clear efficacy within the 25–100 mg/kg dose range, providing an experimental basis for its further development as an adjunctive therapeutic drug for ALI.
This study still has certain limitations. Firstly, the protective effect and molecular mechanism of API have only been validated in mouse models. API undergoes glucuronidation and sulfation processes in the body, 49 and its pharmacokinetic characteristics in humans may differ significantly from those in mice, which could impact the translatability of our research findings. Future research should adopt in vitro experiments using human lung epithelial cells and macrophages, as well as the development of API-based nanoformulations to overcome existing challenges. Secondly, this study did not explore whether API affects other related pathways, which may also be involved in its lung-protective effects. Future studies that reveal the crosstalk between these pathways and the TLR4/MyD88/NF-κB pathway will help to comprehensively understand the multi-target protective mechanisms of API. Thirdly, in addition to species differences in immunity and drug metabolism, the long-term safety of the API, its tissue distribution, and the optimal dosing regimen remain unclear. These factors are of crucial importance for clinical application. Future research may focus on optimizing API formulations, exploring synergistic effects with other anti-inflammatory agents, and its efficacy across additional ALI models. This comprehensive approach will provide a more robust foundation for the clinical translation of the API.
In summary, this study demonstrates that apigenin effectively alleviates LPS-induced acute lung injury by inhibiting the TLR4/MyD88/NF-κB signaling pathway and reducing oxidative stress, providing important preclinical evidence for its potential as a therapeutic agent for ALI.
5. Conclusion
This study demonstrates that apigenin effectively alleviates LPS-induced acute lung injury by suppressing the TLR4/MyD88/NF-κB signalling pathway and reducing oxidative stress. Its protective effect is dose-dependent, with high-dose apigenin (100 mg/kg) exhibiting efficacy comparable to that of dexamethasone. These findings provide important preclinical evidence supporting the potential of apigenin as a therapeutic agent for acute lung injury. Further studies in human-relevant models, focusing on its bioavailability, long-term safety and mechanism of action, will be crucial for advancing its clinical application.
Supplemental Material
Supplemental Material - Apigenin Attenuates Lipopolysaccharide-Induced Acute Lung Injury in Mice by Inhibiting the TLR4/MyD88/NFκB Signaling Pathway
Supplemental Material for Apigenin Attenuates Lipopolysaccharide-Induced Acute Lung Injury in Mice by Inhibiting the TLR4/MyD88/NFκB Signaling Pathway by Haiqiang Chai, Bahman Yousefi and Huixia Liu in Natural Product Communications.
Footnotes
Acknowledgements
The authors would like to thank the staff at Zhinanzhen Biology Laboratory for their technical assistance in animal experiments and histological analysis. We also acknowledge the support from the Department of General Practice, Xi’an International Medical Center Hospital, and the Molecular Medicine Research Center at Tabriz University of Medical Sciences for providing research facilities.
Ethical Considerations
All animal experimental procedures involved in this study were reviewed and approved by Zhinanzhen Biology Ethics Committee (Approval Number: A2024000336). The study was conducted in accordance with the guidelines for the welfare and ethical use of animals and complied with the ARRIVE 2.0 guidelines. 34
Consent to Participate
This study involved animal experiments and no human participants.
Consent for Publication
This study does not contain any identifiable personal data.
Author Contributions
Haiqiang Chai: Conceptualization, Investigation, Data curation, Writing – original draft, Visualization. Bahman Yousefi: Methodology, Formal analysis, Validation, Writing – review & editing. Huixia Liu: Resources, Supervision, Project administration, Writing – review & editing. All authors have read and approved the final manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The datasets generated and analyzed during the current study are available from the corresponding author upon reasonable request. The raw data are not publicly stored in a repository due to the nature of the original experimental records and animal model data.
Supplemental Material
Supplemental material for this article is available online.
Appendix
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.