
Abstract
Spatially and temporally controlled drug delivery is an important field to address the limitations of conventional pharmaceutical administration. While many effective controlled drug delivery systems exist, the repertoire of systems that additionally present a beneficial mechanical environment to cells remains scarce. To address this, a comprehensive release study of fluorescein as a model drug, and the corticosteroid dexamethasone, from poly(N-isopropylacrylamide)/polypyrrole (pNIPAM/PPy) conducting polymer hydrogels is presented within this study. Cyclic voltammetry and scanning electron microscopy (SEM) indicated that having the pNIPAM hydrogel phase present and doping with drugs reduced PPy thickness and shifted/suppressed redox peaks to some degree but not enough to prevent release. Fluorescein release was initiated by constant reduction, with a maximum of 54.5 ± 6.8 µg/cm2 from PPy films and 6.3 ± 1.1 µg/cm2 from pNIPAM/PPy. The quantity of fluorescein released was shown to be tunable by modulating the charge passed during PPy electropolymerization. Fluorescein-loaded pNIPAM/PPy samples were capable of multiple cycles of depletion and reloading via re-incorporation through re-oxidation in a fluorescein solution. The stability of pNIPAM/PPy regarding drug release was demonstrated, with no difference in release profiles and quantities after soaking samples for 1, 8, and 15 days. Interestingly, constant reduction did not elicit release of dexamethasone, while a biphasic pulsed potential of ±0.8 V at 0.5 Hz was effective. Minimal leaching of dexamethasone without stimulation was shown, alongside a multi-day, multi-triggerable release profile upon short stimulations. pNIPAM/PPy conducting polymer hydrogels are a promising platform for on/off drug delivery, with a nondegrading matrix, minimal passive drug-leaching, and where the drug payload can be reloaded, all while providing a suitable mechanical environment to interface with living cells.
Impact Statement
This work demonstrates the inclusion and release of small-molecule drugs from conducting polymer hydrogels, in an electrically triggered manner. This enables the release of drugs to cells in cell culture models, while ensuring a suitable mechanical microenvironment via the hydrogel component. The ability to reload the conducting polymer hydrogel with drug after release is demonstrated, along with excellent control over drug release, with minimal release without an electrical trigger.
Introduction
The majority of pharmaceutical drugs are formulated in conventional delivery systems, but this leads to challenges in optimal delivery, including poor drug solubility, rapid degradation or clearance, unfavorable pharmacokinetics, and indirect administration. 1 Therefore, improvements are constantly being sought to control the delivery of drugs, making them more effective, efficient, and localized, which also has the benefit of reducing side effects. This is especially relevant within the biomaterials field, where drugs may be delivered directly from an implant material or device. Generally, this involves the development of novel drug delivery systems (DDSs) with spatially and temporally controlled release for efficient dosages and localization of release.
DDSs come in many forms, from nanocarrier systems based on polymeric,2–4 biomimetic,5–7 or inorganic materials,8–11 to stimuli-responsive systems12–14 reacting to cues such as pH, redox triggering, enzymes, temperature, light, ultrasounds, or electrical and magnetic fields, to implantable, 15 oral, 16 or transdermal systems, 15 just to name a few. Extensive detail on the mechanisms, advantages, and challenges of each of these systems is out of the scope of this article; however, excellent reviews exist covering these systems in detail.2,4–6,12,13,17,18 Broadly, many DDSs suffer from challenges such as toxicity, stability, spatiotemporal control, biocompatibility, release responsiveness, and particle accumulation.17–19 Conducting polymer hydrogels (CPHs) are a promising emerging class of material that has the potential to address some of these challenges when designing effective DDSs. Both constituents of CPHs—being hydrogels and conducting polymers (CPs)—have been extensively explored individually as DDSs. The details of these systems as DDSs and how combining their properties can address not only the shortcomings of hydrogel and CP systems but also many shortcomings of DDSs as a whole are outlined in the following sections.
Hydrogels are an extensively researched material for application within many facets of tissue engineering. These three-dimensional, water-swollen networks mimic the natural extracellular matrix and provide a supportive environment for cell growth and tissue development. Hydrogels can be engineered to possess tunable mechanical and biochemical properties, allowing researchers to tailor them to specific tissue types and applications. The physical properties of hydrogels can be tailored using a wide range of nano- and microfabrication methods,20–22 while the bulk mechanical properties of stiffness,23–25 porosity,26,27 and degradation properties 28 are also commonly controlled. Hydrogels have also been extensively utilized for biochemically influenced cell studies, where specific signaling molecules are incorporated in the gels. The type of cargo encapsulated within the hydrogels, and the means through which they are released, depends heavily on properties of the hydrogel that can be either tuned toward or selected for, such as the type of gelation (physical, cross-linking, etc), the density and mesh size of the hydrogel, micro and nano structures of the hydrogel, and certain functional groups that could physically or chemically interact with the cargo. 29 The range of these properties that are achievable has made hydrogels viable as DDSs for molecules ranging from small drugs30–34 to proteins and growth factors. 29 Drug release from hydrogels is naturally a diffusion-based process—the kinetics of which depend on the hydrogel mesh size.35–37 Modifications can be made to better control this release, though, such as utilizing hydrogel degradation,37,38 covalent bonding and subsequent decoupling, 39 or physical bonding. 40
CPs have also seen extensive use within drug delivery, although their application commonly extends to flexible electronics, 41 energy storage, 42 sensors, 43 and actuators. 44 CPs have found particular use as DDSs owing to their potential for electrically controlled movement of charged molecules. Controlled release DDSs offer a range of advantages over traditional therapies by maintaining drug concentrations at ideal levels, releasing at desired intervals, and being able to release directly to the site. 45 This controlled release of bioactive molecules from CPs was first reported in the 1980s, 46 and recent effort has centred around improving the controlled characteristics of this release47,48 and applying CP DDSs in clinical trials. 49 Polypyrrole (PPy) is the most common CP used for drug delivery,50,51 although PEDOT and PANI are also well-researched options.52–54 Despite the control that CP DDSs offer, one major drawback when considering cell culture or clinical studies is the relatively rigid nature of the films, which does not match the mechanical properties of any tissues, therefore, creating an unideal mechanical microenvironment for most cells.
CPHs have demonstrated versatile implementation in a range of fields such as energy storage55,56 and sensing. 57 Within the context of drug delivery, though, the tissue-like mechanical properties, biocompatibility, and electrochemical activity of CPHs make them excellent candidates as DDSs that have to potential to address many of the aforementioned challenges facing many DDSs.58,59 Despite this, the pool of research utilizing CPHs as DDSs is limited, and most systems utilize chemical polymerization of the CP within the hydrogel, which is known to have reduced electrochemical function. 60 Systems with an electrochemically grown CP phase for improved electroresponsive behavior, and hence control over release, have been scarcely explored. 59 Examples of this include a poly(dimethylacrylamide-co-4-methacryloyloxy benzophenone (5%)-co-4-styrenesulfonate (2.5%)/poly(3,4-ethylenedioxythiophene) (PDMAAp/PEDOT) CPH system used to release dexamethasone with a −0.5 V constant potential, 61 a gelatin methacryloyl/poly(3,4-ethylenedioxythiophene) (GelMA/PEDOT) CPH which released BSA with −0.6 V constant potential and a biphasic pulse, 62 and a gelatin methacryloyl/polypyrrole (GelMA/PPy) CPH, which released glutamate via constant reduction at −0.6V. 63
This study explores a pNIPAM/PPy CPH system as a new means of controlled delivery of bioactive molecules to cells. This expands the currently lacking space of electrochemically grown DDSs and demonstrates a system with increased unstimulated drug retention, electrochemical responsiveness, and biocompatibility relative to the current repertoire of CP and CPH-based DDSs. Specifically, this article aims to characterize the passive and electrically controlled release mechanisms of fluorescein, as a model drug, and dexamethasone, as a therapeutic drug, from these pNIPAM/PPy CPHs. Fluorescein has been commonly utilized when characterizing the release properties of many DDSs due to its ease of quantification and similar charge, size, and aromatic structure to many therapeutic drugs. Dexamethasone is a corticosteroid that was used in the form dexamethasone 21-phosphate disodium salt (Dex) for a range of reasons. First, Dex and fluorescein are similarly sized (MwDex = 516.4 g/mol and Mwfluorescein = 376.3 g/mol), which is significant as incorporating large molecules into CPs is difficult, 47 although it has been performed before. 62 Both molecules are also dianionic, which is important considering that charge is the driving force for both loading and release. The structures of Dex and fluorescein are also similar, as they both consist of many cyclic carbon groups. Dex has also been used extensively throughout literature as an electrically releasable drug from CPs,64–66 although it has only been demonstrated once for electrochemically synthesized CPH systems (PDMAAP/PEDOT). 61 Previously within our research group, the mechanical properties 67 and cytocompatibility 68 of this pNIPAM/PPy system was studied which, in summary, demonstrated cytocompatibility while maintaining structural integrity, as well as a Young’s modulus of 15.21 ± 0.91 kPa (for the gel composition used in this article) that can be adjusted to as low as 0.66 ± 0.08 kPa by modulating the monomer:crosslinker ratio.
The system was characterized by cyclic voltammetry (CV) to determine the impact of drug loading on redox function, and scanning electron microscopy (SEM) to elucidate any morphological changes when loading fluorescein or Dex into the PPy phase. A comprehensive release study for fluorescein is presented, which includes characterization of release when PPy is in different redox states, the ability to tune the magnitude of drug release with PPy thickness, the stability of release over time, and the ability to reload and rerelease fluorescein. Finally, this work demonstrates temporally controllable, electrically triggered release of dexamethasone from pNIPAM/PPy. An overview of the key mechanisms and results from this study is outlined in Figure 1.
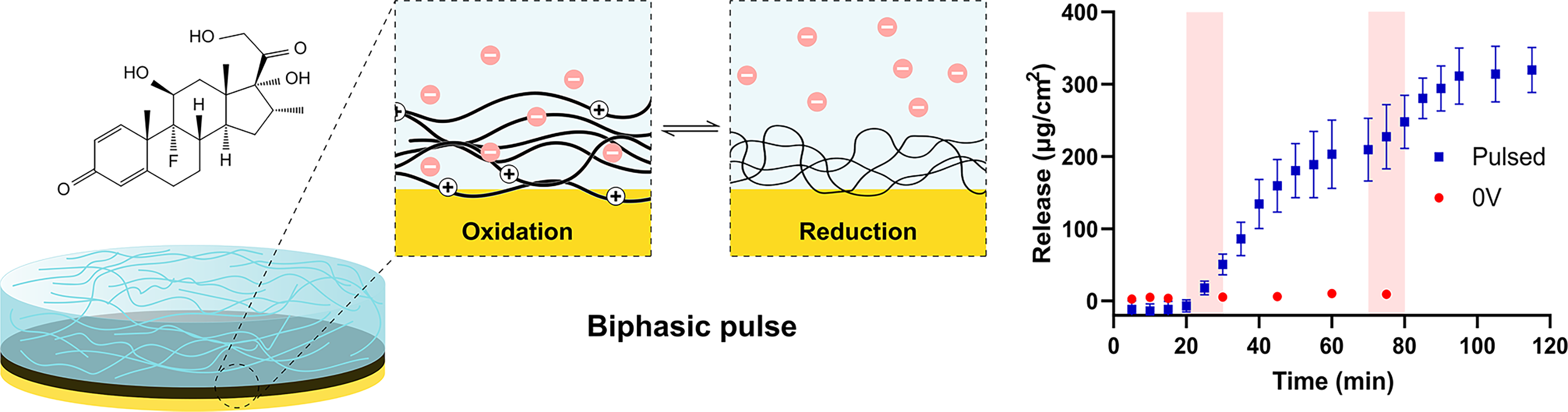
Summary of the key mechanisms and findings of the article.
Materials and Methods
Materials
Hydrogen peroxide (30%, 1L stabilized), N,N′-methylenebis(acrylamide) (BIS, 99%), N,N, N′,N′-tetramethylethane-1,2-diamine (TEMED, 99%), ammonium persulphate (APS, 98%), pyrrole (98%), sodium dodecylbenzene sulphonate (DBS), glutaraldehyde (∼50% in H2O), dichlorodimethylsilane (≥99.5%), sodium chloride (≥99.5%, AT), and fluorescein sodium salt were purchased from Sigma Aldrich, New Zealand. Phosphate-buffered saline (PBS tablets, 5 g, pH 7.45) was purchased from Gibco, Thermo Fisher Scientific (Auckland, New Zealand). 11-amino-1-undecanethiol hydrochloride (99%) was purchased from Prochimia Surfaces, Poland. N-isopropylacrylamide (NIPAM, Mequinol stabilized 99%) was purchased from Fisher Scientific, USA. Dexamethasone sodium phosphate (95%) (Dex) was purchased from AK Scientific Inc, USA. Platinum mesh (0.12 mm NA, 0.04 mm wire diameter, 50 × 50 mm, 99% purity) was purchased from GoodFellow, United Kingdom. Ag/AgCl reference electrode (3M NaCl) and Ag wire (12-inch) were purchased from BASi, USA. Gold-coated silicon wafers (100 mm, P-type/Boron, <100>, 380 µm, 10 nm Ti adhesion layer, 100 nm Au) were purchased from University Wafer, USA. Sulphuric acid (98%, AR) was purchased from LOBA Chemie, India. Ninety-six-well PS F-bottom microplates (Chimney well, solid clear bottom, black, nonbinding) were purchased from Lab Supply Limited.
Fabrication of PPy films and pNIPAM/PPy CPHs
Substrate preparation
Substrates were prepared in accordance with protocols previously established within the group.68,69 In short, gold-coated silicon wafers were cleaned using acid piranha treatment and then rinsed with type 1 water. This was the extent of substrate preparation for samples without pNIPAM and also for pNIPAM/PPy/Dex samples (where PPy was grown before pNIPAM). For pNIPAM/PPy and pNIPAM/PPy/fluorescein samples, the cleaned wafers were soaked in 1 mM 11-amino-1-undecanethiol hydrochloride in an AR ethanol solution for 2 h for amine functionalization, then soaked in a 0.1% (v/v) glutaraldehyde solution in PBS for 30 min.
For samples involving a pNIPAM hydrogel phase, a 200-µm parafilm piece slightly larger than the wafer dimensions was placed across the back silicon face of the wafer, with ∼8 mm excess parafilm being folded back over and pressed flush with the gold surface of the wafer. For this study, wafer pieces were 8 × 8 mm prescored, and eventually cut to 8 × 24 mm. See Supplementary Figure S1 for annotated photographs of this process.
Hydrogel fabrication
pNIPAM hydrogels were also prepared as described in past work.67–69 Briefly, to make a ∼1 mL hydrogel precursor, 850 µL of 0.59 M NIPAM solution (such that final [NIPAM] = 0.5 M), 150 µL of 2% BIS, 13.5 µL of 20% TEMED (v/v), and 20 µL 10% APS—all prepared in deoxygenated Type 1 water, were mixed together. Acting quickly to prevent premature gelation, a volume of hydrogel precursor solution was pipetted onto a DCMS-coated hydrophobic quartz sheet, and the treated wafers were carefully placed gold-side down on top. Once polymerized, the parafilm was removed, and the gels were rinsed in alternating cold and warm water three times to remove unreacted monomer. Hydrogels were then dehydrated, where they can be cut to size for experiments. See Supplementary Figure S1 for annotated photographs of this process.
CP and CPH fabrication
For incorporation of the CP phase, gels were reswelled for ∼16 h 0.1 M NaDBS and 0.1 M pyrrole in type 1 water adjusted to pH 6. The pyrrole/DBS swollen gels were then oxidized using a three-electrode setup, with the 8 × 24 mm gold-coated hydrogel-bound silicon wafers as the working electrode (clipping onto the gold exposed portion), platinum mesh as the counter electrode, and an aqueous Ag/AgCl reference electrode. This electropolymerization and future release studies were performed using a PalmSens3 potentiostat (Palmsens, Netherlands) on the PSTrace version 5.8 software. A constant voltage of +0.7V was held until 0.6 C/cm2 was passed with 300 rpm stirring. PPy samples without the hydrogel phase were made the same way but by simply electropolymerizing onto a clean gold-coated silicon wafer in the pyrrole/DBS solution.
CP and CPH samples with fluorescein were prepared similarly, but 1 mg/mL (2.66 M) fluorescein was dissolved in the pyrrole/DBS solution.
PPy/Dex were prepared with a 0.2 M pyrrole and 0.05M Dex solution without DBS at pH 3. Electropolymerization was performed in the same three-electrode setup by holding a constant current of 2 mA/cm2 for 300 s (0.6 C/cm2 total charge passed). For pNIPAM/PPy/Dex samples, PPy/Dex was made first, and pNIPAM was cast atop according to section 2.2.2.
CPH characterization
CV
CV was performed using the same three-electrode setup as used during electropolymerization. PPy and pNIPAM/PPy hydrogels with a DBS dopant were characterized both with and without fluorescein incorporation. PPy and pNIPAM/PPy were also characterized with Dex incorporation in the absence of DBS. Ten scans were taken in 0.15 M NaCl in Type 1 water within the potential ranges of −1 V to +0.4 V using a scan rate of 0.05 V/s. Data were presented for the 10th scan for each condition.
SEM
pNIPAM/PPy and PPy samples formed on gold-coated silicon wafers were frozen by immersion in liquid nitrogen for 20 s and freeze-dried overnight. The wafers were fractured to expose a cross-section, and samples were loaded onto the SEM analysis puck with carbon tape. An XL30S FEG SEM (Phillips, Netherlands) was used to analyze sample cross-sections with an accelerating voltage of 5.00 kV, spot magnification of 2.0, and magnifications from 70 to 35000 × g. The thickness of PPy, derived from one sample for each condition, was assessed using ImageJ version 1.50i.
UV/vis
The absorbance of Dex was measured between 200 and 350 nm in a Nanodrop (Implen Nanophotometer N50, USA). Two microliters of the solution was used per measurement, and absorbance at 240 nm was taken to determine Dex concentration compared with a standard curve (Supplementary Fig. S2).
Passive release studies
Fluorescein-incorporated and Dex-incorporated pNIPAM/PPy films were constructed using the above methodology. From here, they were rinsed in Type 1 water for 3 s to remove any surface adhered fluorescent or Dex. One 50 mL solution of PBS was made per sample, and with stirring of 300 rpm applied, would act as the release vessel for each sample. Before samples were placed in release vessels, PBS aliquots were collected. Samples were then placed in separate release vessels, and aliquots were taken at each timepoint. The fluorescence of fluorescein samples was then measured via a microplate reader (Perkin Elmer EnSpire Multimode Plate Reader) at Ex/Em = 495/518 nm alongside a fresh standard curve of dissolved fluorescein in PBS ranging from 250 to 7.8 µg/mL. Relative fluorescence units were converted to ng/cm2 released by comparison with the standard curve (Supplementary Fig. S2). Dexamethasone samples were measured with UV/vis by taking absorbance at 240 nm, and conversion to µg/cm2 was conducted via comparison with a standard curve of Dex, ranging from 500 to 7.8 µg/mL. The presented ng/cm2 units denote ng of fluorescein/Dex per cm2 of pNIPAM/PPy on the substrate.
Active release studies
Both PPy CP films and pNIPAM/PPy CPHs were analyzed for electrochemically induced release. For all active release measurements involving pNIPAM, samples were placed in PBS overnight (fluorescein) or for 2 h (Dex) prior to the electrochemical release to ensure passive release from the hydrogel was complete. For PPy films, samples were simply rinsed with type 1 water. The only cargo remaining in the system should now be within the PPy. All active release data are also presented in ng/cm2, which denotes ng of fluorescein/Dex per cm2 of pNIPAM/PPy on the substrate.
Effect of PPy redox state on fluorescein release
To explore the quantities of release, release profiles, differences in release with pNIPAM, and redox states generating release, release experiments exploring different potentials were performed on PPy and pNIPAM/PPy. A three-electrode setup was used with a sample-coated gold working electrode, a platinum mesh counter electrode, an Ag/AgCl reference electrode, and 100 mL PBS as the electrolyte stirred at 800 rpm. A PalmSens 3 potentiostat was used in chronoamperometry to control potential during these experiments. A 100 µL PBS was extracted and placed in a 96-well plate before the three-electrode system was lowered in. After 5 min, another measurement was taken before the potential was applied. Conditions tested for PPy films were −0.6 V (reduction), +0.4 V (oxidation), −0.6 V to +0.4 V pulsing (0.5 Hz), and no voltage. For pNIPAM/PPy samples, conditions tested were −1.0 V (reduction), +0.4 V (oxidation), −1.0 V without PPy, and no voltage. A total of 100 µL of electrolyte was extracted and placed into a 96-well plate every 5 min for a further 30 min, totaling 35 min for PPy films, and every 15 min for another additional 30 min, totaling 65 min for pNIPAM/PPy hydrogels. A microplate reader was used alongside a fresh standard curve to determine fluorescence at each time point.
Effect of PPy thickness on fluorescein release
To investigate the effect of varying charge passed during PPy polymerization, PPy and pNIPAM/PPy films were constructed according to the above procedure, but with 0.2, 0.4, 0.6, 0.8, and 1.0 C/cm2 of charge passed for PPy films, and 0.2, 0.6, and 1.0 C/cm2 for pNIPAM/PPy films. −0.6 V reduction was held when measuring release from PPy films, and −1.0 V was held for pNIPAM/PPy hydrogels. Measurements were taken as described above.
Effect of extended storage on fluorescein release
To determine if fluorescein could be retained in pNIPAM/PPy when stored in an aqueous environment, fluorescein-incorporated pNIPAM/PPy hydrogels were left in PBS for 1, 8, and 15 days before being reduced at −1.0 V for measurement.
Release and reload of fluorescein
Fluorescein-loaded pNIPAM/PPy hydrogels were reduced at −1.0 V, and release was measured as typical. The same samples were then reloaded by oxidation at +0.4 V in a 1 mg/mL fluorescein solution in type 1 water. This oxidation was held until the charge passed equaled the charge of the preceding reduction step. These “reloaded” samples were then reduced again to release, at −1.0 V. This process was repeated once more.
Active control over the release of dexamethasone
Dex release measurements were performed using the same three-electrode setup by applying a biphasic pulsed potential of ±0.8 V at 0.5 Hz in 1.5 mL of PBS solution. The pulsed potential was held for 10 min, except during the long-term, gradual release investigation, where 10 s of pulsed potential was used. A total of 5 µL of solution was removed and stored in an Eppendorf tube for later measurement at each time point. The influence of removing this solution was accounted for when converting the absorbance output to µg/cm2 of Dex release.
Statistical analysis
Results are reported as the mean ± standard deviation. Sample sizes were three or four samples, which are clarified per experiment. A Student’s two-sided t-test was employed to determine statistical significance, with significance accepted at p < 0.05. All statistical calculations and graph plotting were performed using Prism version 8.0.2 (GraphPad Software, San Diego, CA).
Results and Discussion
Electrochemical response with drug loading
CV was utilized to investigate changes in the electrochemical response of PPy when doped with different drugs and when a hydrogel phase is present (Fig. 2). The presented CVs are the tenth CV cycle, which is after the CVs had stabilized.
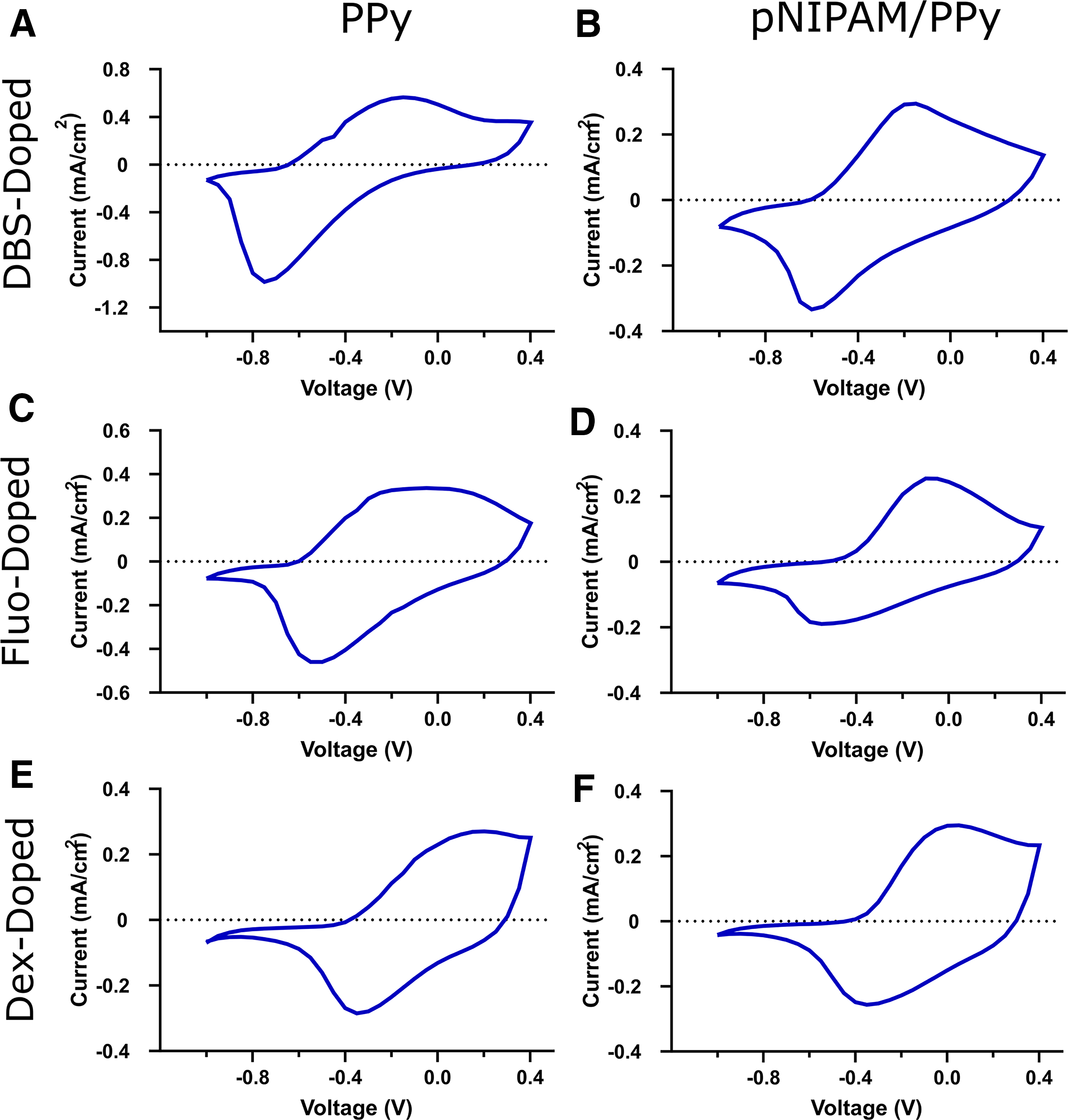
CV between −1.0V and +0.4V at a scan rate of 0.05V/s over 10 scans for
When analyzing the influence of pNIPAM on electrochemical response, it can be noted that the reduction peak current is far greater for PPy samples compared with pNIPAM/PPy. The oxidation peak current was also significantly larger for PPy DBS samples than for pNIPAM/PPy DBS samples. PPy is known for its efficient and relatively reversible redox behavior, and clearly, the hydrogel phase affects the reduction reaction. This could be due to a variety of factors; for instance, the hydrogel phase could influence the morphology of the PPy in such a way that it limits the reduction process, or the hydrogel could be acting as a barrier for the diffusion of ions. Reduction peaks were generally also broader for pNIPAM/PPy compared with PPy, which speaks toward the ion diffusion-limiting nature of the hydrogel. 70 The oxidation and reduction peak positions for PPy samples and their hydrogel counterparts were generally comparable.
When comparing how different drugs as dopants influence electrochemical response, it is observed that both the oxidation and reduction peak currents are at lower magnitudes when fluorescein or Dex are incorporated. This is once again more significant for the reduction peak but also more significant for PPy films than pNIPAM/PPy hydrogels. This is likely due to the different means of ion transport during redox for drug-loaded samples. For DBS-doped samples, during the reduction sweep, the PPy backbone becomes neutral from positive, which introduces a net negative charge due to the presence of negatively charged DBS, and this acts as a driving force for the expulsion of DBS. However, DBS is a relatively large dopant that forms even larger micelles, which remain trapped within the PPy. Instead, Na+ from the electrolyte migrates into the PPy to neutralize this charge. For fluorescein and Dex-doped samples, these dopants are small enough to be expelled from the PPy during reduction. 69 While the reduction and oxidation peak positions for both fluorescein conditions are comparable to solely DBS-doped samples, Dex samples show significant peak shifting. These reduction peaks are located at ∼−0.35 V, and the oxidation peaks are shifted to ∼−0 V. Peak position shifting is commonly observed when using different dopants, 69 and the peaks of fluorescein samples remaining relatively unchanged is likely due to DBS still being present, as fluorescein samples were codoped, while Dex was the sole dopant for Dex samples.
These changes in redox peaks are not unusual when incorporating a hydrogel phase or altering the selected dopant, where peaks have been documented to shift in both position and intensity.52,69,71,72 Despite the differences when introducing pNIPAM and when doping with fluorescein and Dex, it is clear that all conditions tested are electrochemically active with clear redox peaks, which is the first factor when considering whether these systems can be used as DDSs.
Morphological characterization with drug loading
For the goal of incorporating and releasing different bioactive molecules from pNIPAM/PPy, it is important to understand how using these molecules as dopants for PPy influences its morphology. It is also important to evaluate the effects of electropolymerizing PPy through pNIPAM compared with bare gold. For this, SEM cross-sections of PPy and pNIPAM/PPy samples doped with DBS, DBS+fluorescein, and Dex were imaged, as shown in Figure 3. pNIPAM delaminated from the substrates during liquid nitrogen immersion; however, the delaminated pNIPAM was optically clear, indicating no PPy was lost from the substrate. Only Dex/PPy is included, as for Dex release studies, release was only possible when pNIPAM was cast directly onto Dex/PPy films, rather than being electropolymerized through pNIPAM.

SEM cross-section images of PPy and pNIPAM/PPy. pNIPAM was delaminated from the substrate upon liquid nitrogen exposure, leaving the PPy film behind. For pNIPAM/PPy Dex, the hydrogel remained. The PPy cross-section is labeled, and the thickness of each is shown. pNIPAM/PPy, Poly(N-isopropylacrylamide)/polypyrrole.
The PPy layer for PPy films, compared with pNIPAM/PPy films, was significantly thicker in both cases. With DBS as the dopant, PPy thickness decreases from 3.5 µm without pNIPAM to 2.3 µm with pNIPAM (Fig. 3A and B). With DBS and fluorescein as dopants, PPy thickness decreases from 5.8 µm without pNIPAM to 0.9 µm with pNIPAM (Fig. 3C and D). PPy thickness when doped with Dex was 6.0 µm (Fig. 3E). This is likely due to the initial thiolizing step for pNIPAM/PPy samples, as it has been shown through electrochemical impedance spectroscopy studies that thiol layers, and polymer self-assembled layers in general, increase the charge transfer resistance of the substrate.73–75
The PPy film was produced with DBS as the dopant and was found to be thinner compared with those doped with fluorescein or Dex. DBS is a common and established dopant for CP systems, and this observation could be due to films polymerizing in a more densely packed fashion with DBS, compared to more porous films for fluorescein or Dex dopants. It is important to consider that while constant potential was used to construct DBS and DBS + fluorescein samples, constant current was used to electropolymerize Dex samples, due to otherwise slow and inhomogeneous polymerization. The polymerization technique used is known to influence CP properties. 60 Dopant choice appears to influence the magnitude to which the presence of pNIPAM affects PPy thickness. The most significant example is when DBS + fluorescein is doped.
Release of charged molecules from pNIPAM/PPy
Effect of PPy redox state on fluorescein release
The following sections detail the electrically stimulated release of fluorescein from PPy and pNIPAM/PPy. It is important to note, though, that due to how pNIPAM/PPy films are fabricated, fluorescein also initially exists within the swollen pNIPAM. Release from this hydrogel phase is diffusion-mediated and referred to as passive release, while electrically stimulated release from the PPy phase is referred to as active release. Figure 4A details the passive release of fluorescein from pNIPAM/PPy. Fluorescein diffuses rapidly from pNIPAM, with a plateau being reached after 2 h at 122 ± 1.9 µg/cm2 released. Interestingly, the measured amount of fluorescein trends slightly downward over the following days, which could be due to small amounts of fluorescein binding to the container walls, fluorescence degradation, or fluorescein binding to itself. All active release data presented are from samples that have undergone passive release prior to measurement, by soaking overnight in PBS; therefore, active release data are purely electrically stimulated from the PPy, rather than diffusing from the pNIPAM.

Both PPy films and pNIPAM/PPy hydrogels were exposed to different forms of potential application to investigate the ability of electrical stimulation to modulate release. As seen in Figure 4B, which details release from PPy, with a −0.6 V potential applied, 42.1 ± 3.59 µg/cm2 of fluorescein was released within the first 5 min, rising slowly for a further 15 min until reaching a plateau at 54.5 ± 6.83 µg/cm2. No release was detected in the absence of stimulation or when the film was oxidized. When applying a biphasic pulse of −0.6 V to +0.4 V at 0.5 Hz, a gradual release is observed until release significantly slows after 30 min at 14.1 ± 4.45 µg/cm2.
Similar experiments were conducted on samples with the inclusion of the hydrogel phase, pNIPAM/PPy, shown in Figure 4C. The reductive potential chosen was −1.0 V, which is preferred over the −0.6 V used for PPy release because it pushes the system well past the reductive peak (Fig. 2) and ensures the transitioning of the PPy into an electrically neutral reductive state. Without the pNIPAM hydrogel phase, a −1.0 V potential would partly reduce the gold WE, leading to delamination of the PPy from its gold substrate, but with pNIPAM present, this issue is not seen. When reduced, pNIPAM/PPy films release fluorescein until a plateau after 30 min of 6.31 ± 1.10 µg/cm2. When oxidized at +0.4 V, there was a small but consistent increase in release over the measured 60 min, with a final release of 2.93 ± 2.13 µg/cm2. This is surprising and is not what was observed without the hydrogel phase present, as triggering the PPy to an oxidative state should turn the polymer backbone positive, hence utilizing the negative charge of the DBS dopant and fluorescein to balance these charges and introducing a driving force for maintaining fluorescein within the CP. The two controls for this experiment, being with no voltage applied and when pNIPAM hydrogels were reduced at −1.0 V without the PPy phase, showed an insignificant release. Redox pulsing is not shown for pNIPAM/PPy hydrogels as it was for PPy films, as it was far less efficient for release than reduction for this system.
The mechanism responsible for driving the release of fluorescein is the change in charge of the PPy polymer backbone due to electrical stimulation. When oxidized, the PPy backbone is positive, requiring a negative dopant such as the DBS and fluorescein to balance the charge. When reduced, the backbone shifts to a neutral state, which, considering the incorporation of negative dopants, yields an overall negative charge in the system. DBS is a relatively large micelle-forming dopant and is not expelled from the PPy upon reduction in attempts to neutralize this charge. Instead, positive ions within the electrolyte, such as sodium ions in this case, will traverse into the PPy to balance charges, 76 while simultaneously, the dianionic fluorescein will be expelled, resulting in the observed release. With this mechanism in mind, the comparison between release from PPy with and without the pNIPAM hydrogel phase can be understood. First, PPy films alone have a much greater release relative to pNIPAM/PPy hydrogels, with the plateaued release values for each of these, when reduced, being 54.5 ± 6.83 and 6.31 ± 1.10 µg/cm2, respectively. This can be explained by Figure 4A and D, where PPy films were significantly thinner for pNIPAM/PPy compared to PPy alone. Thinner films should correspond to less fluorescein incorporated. Alongside the increased release of PPy compared with pNIPAM/PPy, these samples also release quickly than pNIPAM/PPy samples. Critically for future applications of this system in cell culture studies, both quantities of fluorescein released are well in excess of what is biologically relevant for many therapeutic drugs.
Effect of PPy thickness on fluorescein release
The ability to tune the quantity of cargo released will be an important quality of this CPH system for future cell studies. Figure 5 presents the release profiles from PPy and pNIPAM/PPy upon −0.6 V reduction and −1.0 V reduction from the films, respectively. The amount of charge passed during electropolymerization while constructing the CP phase was varied from 0.2 to 1.0 C/cm2, where more charge passed corresponds to a thicker PPy layer, and theoretically, higher loading. A clear trend is seen for PPy films in Figure 5A, wherein fluorescein release from 0.2 C/cm2 films reaches a peak at 10.5 ± 6.68 µg/cm2, which was the lowest by a large margin. This was followed by 0.4 and 0.6 C/cm2, which are not significantly different from one another, reaching maximum releases of 50.7 ± 10.8 and 54.6 ± 7.17 µg/cm2, respectively. This was followed by 0.8, then 1.0 C/cm2, releasing 75.8 ± 2.15 and 88.1 ± 0.86 µg/cm2, respectively. Interestingly, for release from the two thickest films, 0.8 and 1.0 C/cm2 films, both seemed not to plateau after 30 min of release as we have come to expect from PPy films. This makes sense because thicker films hold more cargo and hence require a longer time to expel it to completion. The inverse can be seen with 0.2 C/cm2 films, with the plateau being reached after just the initial 5-min measurement. This clear trend of thicker PPy films with more charge passed, releasing more fluorescein is not as pronounced for pNIPAM/PPy hydrogels; 0.2 C/cm2 films released a maximum of 6.98 ± 0.33 µg/cm2 of fluorescein, which is much greater than anticipated when observing PPy release trends in Figure 5A; 0.6 C/cm2 samples released slightly less, at 4.89 ± 0.04 µg/cm2. This could partly be due to sample variance, as it is known that release systems are difficult to replicate perfectly. Thicker samples made by passing 1.0 C/cm2; however, support the hypothesis that thicker films release more cargo, with a maximum release of 12.2 ± 4.1 µg/cm2. This is significantly more than any release observed across all experiments using pNIPAM/PPy.

Electrically triggered fluorescein release profiles as a function of charge passed during electropolymerization of PPy for
Release and reload of fluorescein
The ability for pNIPAM/PPy films to be reloaded with fluorescein is investigated in Figure 5C. This entails reducing samples and measuring their maximum release, which is the quantity of fluorescein released at the plateau. After expelling fluorescein totally, samples can then be reoxidized in a fluorescein solution. This causes the PPy backbone to actuate to a positive state, causing fluorescein anions to migrate back into the backbone to neutralize this charge. The initial reduction released 4.46 ± 1.22 µg/cm2 of fluorescein, while after one reload and release cycle, 4.66 ± 0.4 µg/cm2 was released. These quantities are not significantly different and demonstrate the system’s ability to be reloaded. After a second reload, the fluorescein released is roughly halved to 2.63 ± 0.59 µg/cm2, showing that although this system can be recharged multiple times, this is a finite process. Only two reload cycles were performed due to the fact that the time taken to pass the charge required, when oxidizing in fluorescein, increased significantly for each reload (data not shown). These diminishing properties for subsequent reloads could be due to, first, the compromise of the electrochemical response of the CP for each redox cycle due to degradation, which is well documented when CPs are exposed to CV. 51 Another contributing factor could be that during reduction, the PPy backbone becomes neutral, causing fluorescein to be released while DBS remains trapped. This leads to an overall negative charge, which will be balanced by positive ions, such as sodium, within the electrolyte. Upon further reloads, the positive ions present could dampen the driving force of fluorescein reincorporation.
Effect of extended storage on fluorescein release
A parameter of paramount importance in cell studies is substrate stability. Particularly for this pNIPAM/PPy system, wherein samples would have to stay soaking in water for extended periods to remove toxic monomers prior to cell studies, the ability for the hydrogel to maintain its physical properties and structure, and for the CP to maintain its electrically actuating properties and withhold its cargo is vital. Cell studies and clinical studies with DDSs are also often conducted over weeks, further exemplifying the need for a stable system. This pNIPAM/PPy system without cargo has been used before in cell culture, 68 but it is important to investigate the stability of release, too. Figure 5D depicts release experiments wherein samples were reduced at −1.0 V after being stored in 0.01 M PBS for varied lengths of time. Release after storage for 1, 8, and 15 days was extremely similar, with no significant differences noted. The release values attained after 60 min of applied potential were 4.48 ± 1.22, 4.09 ± 0.10, and 3.84 ± 0.22 µg/cm2 for 1, 8, and 15 days, respectively.
Dexamethasone sodium phosphate release
In terms of fabrication and release, Dex behaved differently than expected compared to fluorescein. For instance, while fluorescein was able to be codoped with DBS at relatively low concentrations (1 mg/mL), Dex/PPy systems in literature use Dex as the sole dopant, and codoping trials did not yield electrically triggered release. This could be due to Dex being encapsulated within the DBS micelles77,78 due to its hydrophobic steroidal core, rendering it trapped within the PPy alongside the DBS micelles, which are already too large to partake in ion migration during redox. Because of this, PPy-Dex films were electropolymerized with Dex as the sole dopant, requiring a much greater concentration than used for fluorescein. Due to the absence of a typical dopant, PPy electropolymerization was also compromised—building less homogeneous films over much longer times. To alleviate this, chronopotentiometry was utilized rather than chronoamperometry, although still passing a total charge of 0.6 C/cm2. Constant potential was shown to be the most effective potential profile to generate the release of fluorescein, as shown in Figure 4B and C. Despite this, most literature releasing Dex from PPy utilize CV,51,64,79 although reports have been presented which use constant potential 66 or pulsed potential. 80 During trials with this system, constant potential and CV were ineffective for electrically stimulating release, while pulsed potential succeeded. This could be due to the absence of DBS, resulting in a less ordered and denser PPy matrix, hence increasing the steric hindrance for the Dex to migrate out during application of a constant potential. Because of this, a more mechanical-based release method may be better suited, like a pulsed potential, wherein instead of release being governed solely by electrostatic forces, the Dex is pumped out by the expansion and contraction of the PPy matrix (Fig. 6). A summary of dopant, fabrication, and release conditions tested are presented in Supplementary Table S1.
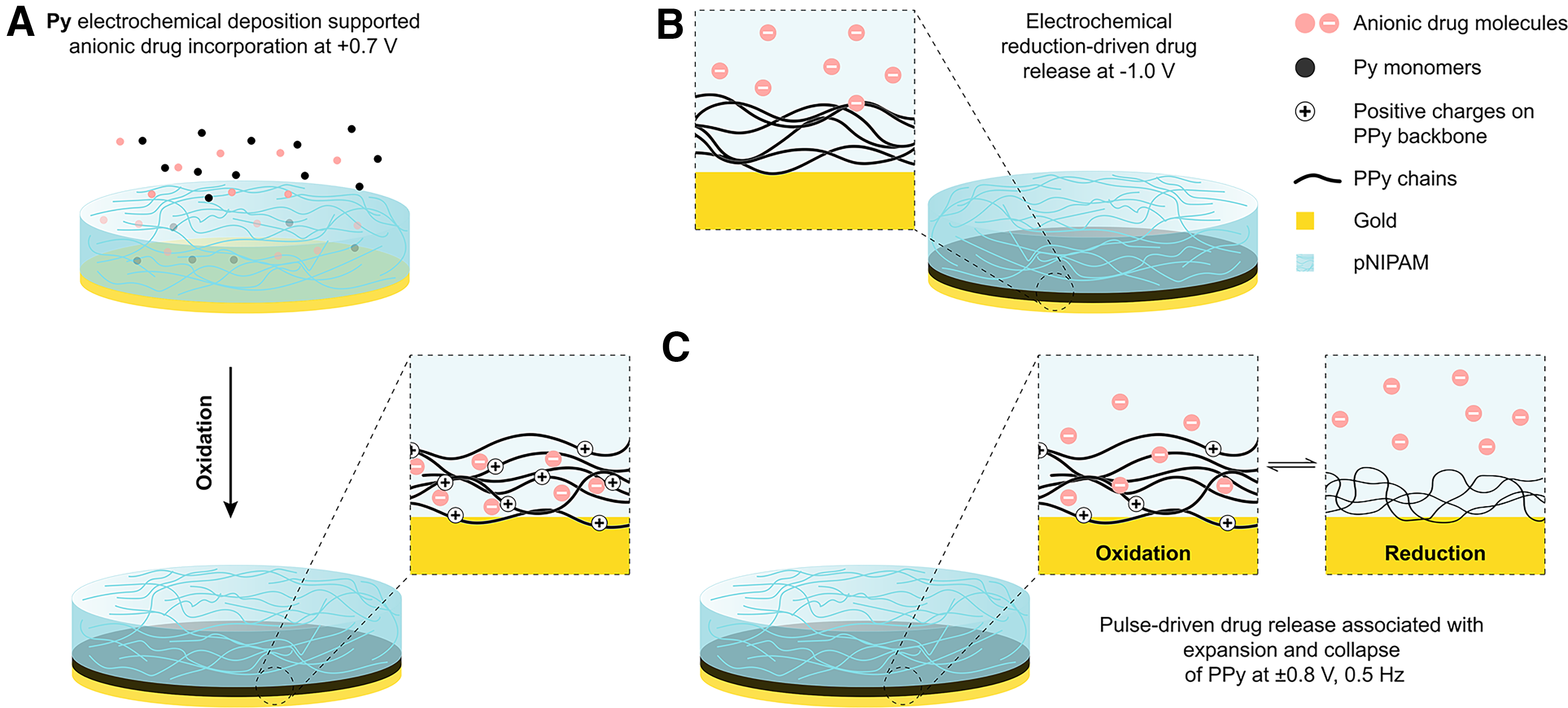
Illustration of the key differences in release mechanisms between fluorescein via constant potential and Dex via pulsed potential.
The electrically controlled release of Dex from PPy is shown in Figure 7A. The potential applied was a pulsed potential at ±0.8 V at 0.5 Hz, which was adapted from past literature.65,80,81 No significant release of Dex was observed within the first 20 min, where no potential was applied for either condition, and for control samples, this continued over the duration of measurement. This is contrary to what can be seen within literature, where Dex release from PPy shows significant passive release of Dex, and active release from electrical stimulation only accelerates, as opposed to commences, Dex release.80,82 When a biphasic pulse is applied between 10 and 20 min, release of Dex is immediately observed. This release continues at a constant rate for a further 15 min after the potential is removed, to 160 ± 36.6 µg/cm2, then slows to a plateau over the following 25 min to a released quantity of 209 ± 43.3 µg/cm2. A second burst of Dex release was then able to be stimulated through a second application of the biphasic pulse at 75 min. Once more, release continued after the removal of applied potential, although a plateau was now reached after 15 min, yielding a final released quantity of 320 ± 31 µg/cm2.
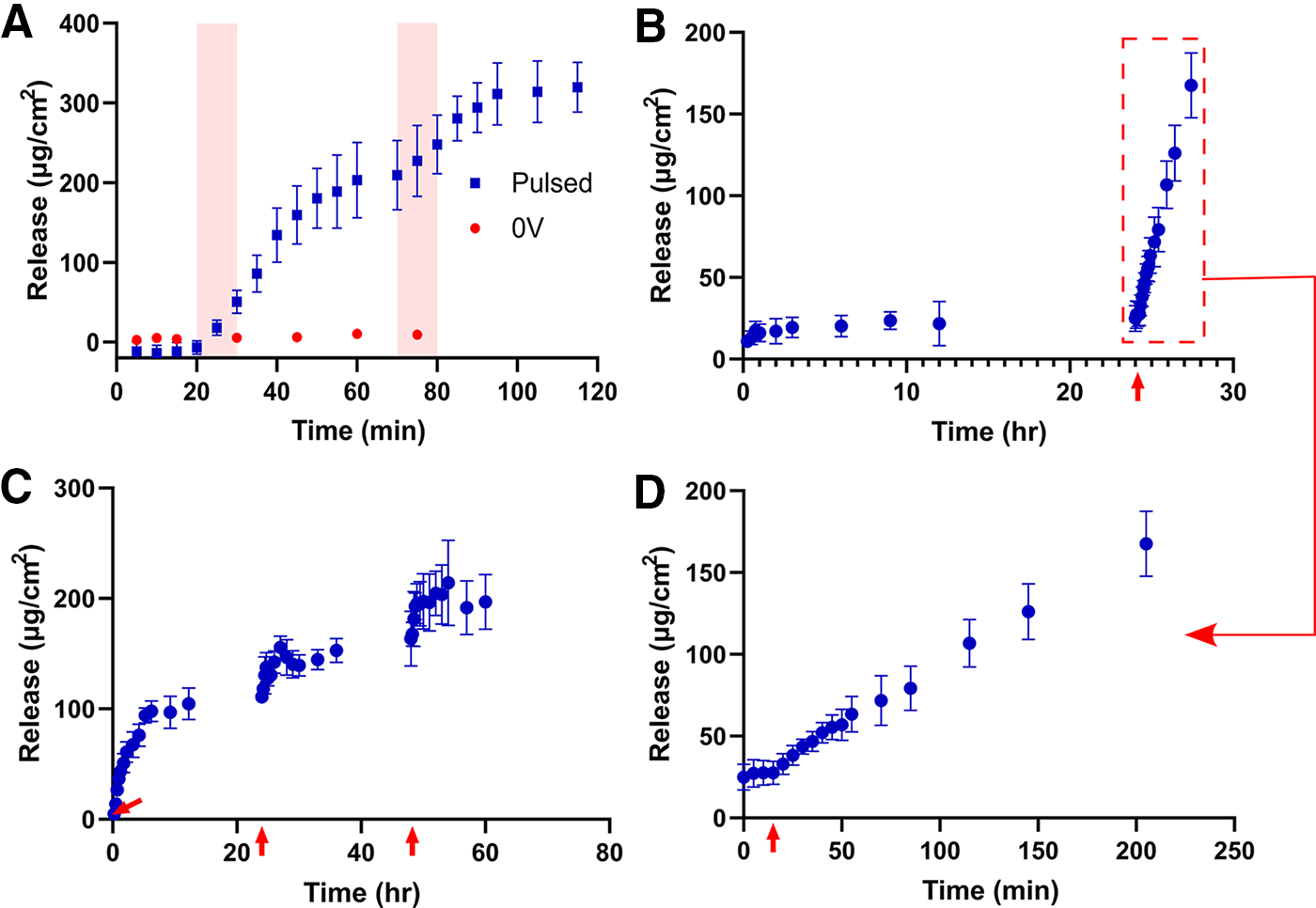
Interestingly, when attempting to achieve triggered release of Dex from pNIPAM/PPy, the typical growth order of pNIPAM gelation first, followed by PPy/Dex incorporation, did not yield the desired triggered release. Instead, by first growing the PPy/Dex, which is shown to be capable of triggered Dex release (Fig. 7A), then gelating pNIPAM on top, release was achieved, as depicted in Figure 7B to D. pNIPAM was still able to adhere to the substrate despite the inability to thiolize pNIPAM to gold in this case. In Figures 7B and D, samples were soaked within PBS for 24 h prior to stimulation, and although an initial release is detected over the first 45 min up to 17.3 ± 5.71 µg/cm2, only a minor subsequent increase in this release was detected over the following 23 h, up to 25.0 ± 7.9 µg/cm2. This minor passive release, as opposed to a completely insignificant passive release without pNIPAM (Fig. 7A), is interesting considering the hydrogel was not swollen in Dex, as was the case for fluorescein samples, and instead was simply cast atop PPy/Dex. Nevertheless, this passive release is removable, considering it does not continuously increase. After 24 h, the same 10-min biphasic pulse was applied to stimulate the samples. Subsequent burst release was observed, yielding a final quantity of 168 ± 19.8 µg/cm2 after 200 min with no indication of plateauing. This is a notably more gradual release relative to when pNIPAM is not present—with this difference being far more pronounced than for fluorescein release. Figure 7C depicts the capabilities for temporally controllable Dex release over long periods of time. The goal of this long-term release study was to better emulate a release profile that would be viable in future cell studies, and therefore, the pulse time was reduced to 10 s to slow down the release and to increase the chance of cells surviving stimulation. Three pulses were applied to the samples, 24 h apart. The release of Dex from the initial pulsing was the greatest, plateauing after 5 h to 94.2 ± 6.9 µg/cm2. The following two pulses stimulated further release, up to maximums of 156 ± 10.3 and 214 ± 24.7 µg/cm2, respectively. The ability for multiple triggered release events to occur with this system is an important quality when envisioning the system’s potential use in cell culture, as both a spatially and temporally controllable release profile is important.
This study demonstrates a promising foundation for future in vivo applications by addressing key challenges commonly encountered in DDSs. The system’s stability over extended periods suggests favorable long-term performance, a critical requirement for in vivo deployment. While biological complexity introduces variability, the tunable properties of this delivery platform allow for potential adaptation to physiological conditions. Together, these characteristics underscore the translational potential of the system and warrant further investigation in cell studies, eventually, preclinical trials.
Conclusion
The ability of pNIPAM/PPy CPHs to incorporate and release bioactive molecules was explored in this study. First, the electroresponsive properties of PPy and pNIPAM/PPy were explored using CV for samples prepared with and without fluorescein and Dex. While the general shape of CV curves consistently demonstrated a reversibly electroactive material, there were differences that arose from fluorescein and Dex incorporation, such as peak shifting and lowering in the current at said peaks. Critically though, PPy and pNIPAM/PPy were both found to still be significantly electrically responsive with incorporation of fluorescein and Dex. SEM demonstrated how the presence of pNIPAM reduces the thickness of the underlying PPy layer and that this is most extreme with fluorescein as a dopant. Successful fluorescein release was achieved from PPy and pNIPAM/PPy using a constant reducing potential. It was shown that release quantities could be modulated by adjusting the current passed during the electropolymerization of PPy. It was also shown that pNIPAM/PPy could be reloaded with fluorescein after releasing, although the efficacy of reloading was diminished with increasing release-reload cycles. The stability of the system was demonstrated with pNIPAM/PPy soaking for 1, 8, and 15 days all releasing the same quantities of fluorescein. Finally, Dex was shown to release from both PPy and pNIPAM/PPy films with a biphasic pulse as the trigger mechanism, and long-term release with minimal passive release or leaching of Dex was demonstrated. This system holds great promise as an electrically actuatable DDS with favorable mechanical properties for cell culture.
Authors’ Contributions
M.S.H. contributed to conceptualization (equal), methodology (lead), investigation (lead), writing—original draft (lead), and visualization (lead). K.E.Z. contributed to conceptualization (equal), methodology (supporting), investigation (supporting), writing—review and editing, and visualization (supporting). M.S.T. contributed to conceptualization (equal), methodology (supporting), and writing—review and editing. D.S. contributed to conceptualization (supporting), writing—review and editing, and supervision (supporting). J.M. contributed to conceptualization (equal), resources, writing—review and editing, supervision (lead), project administration, and funding acquisition.
Footnotes
Acknowledgments
The authors gratefully acknowledge the financial support through the Rutherford Discovery Fellowship, from Government funding, managed by Royal Society Te Apārangi, and from the MacDiarmid Institute for Advanced Materials and Nanotechnology. The authors also acknowledge Catherine Horris for her assistance with SEM.
Funding Information
No funding was received for this article.
Disclosure Statement
No competing financial interests exist.
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.