
Abstract
Conventional drug screening models face a series of challenges in guiding individualized cancer treatment, including time-consuming processes, limited drug coverage, and insufficient accuracy in efficacy evaluation. This study aims to establish a convenient, rapid, and reliable drug screening protocol for evaluating individualized efficacy of chemotherapy and immunotherapy. We established an ex vivo mini-tumor culture platform by culturing tumor fragments in an air–liquid interface system, which was capable of sustaining tumor growth for at least 2 weeks and maintaining immune cell infiltration for over 1 week. Using this mini-tumor culture platform, we can evaluate the individualized therapeutic responses of different tumors to chemotherapy and immunotherapy, including gemcitabine, 5-fluorouracil, cisplatin, αPD-1 and αPD-L1. Furthermore, using this drug evaluation platform, we revealed distinct therapeutic responses to immunotherapy between immune-cold tumors and immune-hot tumors, and demonstrated the important role of the immunologic adjuvant resiquimod (R848) in enhancing immunotherapy efficacy. This mini-tumor culture protocol provides a feasible implementation approach for ex vivo personalized drug testing.
Impact Statement
In this study, we have established an ex vivo mini-tumor culture platform capable of sustaining tumor growth for at least 2 weeks and maintaining immune cell infiltration for 1 week, providing a sufficient time window for drug sensitivity testing. Using this mini-tumor culture platform, we can evaluate the individualized therapeutic responses of different tumors to chemotherapy and immunotherapy, including gemcitabine, 5-fluorouracil, cisplatin, αPD-1 and αPD-L1. Furthermore, using this drug evaluation platform, we revealed distinct therapeutic responses to immunotherapy between immune-cold tumors and immune-hot tumors, and demonstrated the important role of the immunologic adjuvant resiquimod (R848) in enhancing immunotherapy efficacy. This mini-tumor culture protocol provides a feasible implementation approach for ex vivo personalized drug testing.
Introduction
Cancer ranks among the leading causes of death globally. In 2022, the world recorded 20 million new cancer cases and 9.7 million cancer-related deaths. 1 According to the World Health Organization, it is estimated that by 2050, the number of annual new cancer cases will exceed 35 million worldwide, an increase of 77% compared with 2022. Drug therapy remains the primary treatment option for cancer therapy, with traditional chemotherapy and emerging immunotherapy still widely used clinically. However, drug resistance is a significant characteristic of many cancer types and continues to pose a major obstacle to achieving a cure for cancer patients. 2 However, due to tumor heterogeneity, fractions of tumor cells survived from initial therapy, often leading to treatment failure and tumor recurrence. 3 Recurrent cancers typically carry a poor prognosis. Therefore, developing personalized approach is a promising strategy to improve the accuracy of cancer treatment and reduce cancer recurrence rates.
In recent years, significant progress has been made in tumor immunotherapy. At present, the most widely used and successful immunotherapy in clinical practice is the immune checkpoint inhibitor (ICI) therapy. Immune checkpoint therapy has been approved by the FDA for the treatment of a variety of cancers. 4 Since ipilimumab was approved for the treatment of metastatic melanoma in 2011, three classes of ICI therapies (targeting PD-1, PD-L1, LAG-3) have received FDA approval for human cancer treatment. Although ICIs have demonstrated remarkable efficacy in certain cancers, many patients and cancer types still respond poorly to ICI treatment. For instance, Pembrolizumab demonstrated varying objective response rates (ORR) across cancer types: ovarian cancer, ORR of ∼24% 5 ; head and neck cancer, ORR of ∼17% 6 ; nonsmall cell lung cancer, ORR of ∼40% 7 ; metastatic urothelial carcinoma, ORR of ∼28.6%. 8 In addition, while exerting antitumor effects, ICI may nonspecifically activate the immune system, damaging normal organs. Immune-related adverse events typically occur within the first few weeks to months after treatment. 9 Therefore, there is an urgent need to evaluate ICI drug efficacy using ex vivo models prior to actual clinical treatment.
To address the demand for individualized cancer therapy, many ex vivo model systems have been developed, including two-dimensional (2D) primary cell line culture, 10 patient-derived xenograft (PDX), 11 genetically engineered mouse models (GEMMs), 12 and three-dimension (3D) cell culture model. 13 However, these models still have their own limitations. For example, 2D culture fails to mimic the natural structures of tumor tissues and lacks tumor heterogeneity, and its drug response data often show discordance with therapeutic efficacy observed in clinic. 14 Animal models including PDX and GEMMs, while preserving the complex biology of patient tumors, require substantial time and financial investment to establish. 15 In contrast, 3D cell culture demonstrated numerous advantages. In recent years, 3D cell culture has developed rapidly. Various models such as organoids, spheroids and tissue explants have been successfully developed and applied. 3D cell culture techniques are regarded as a tool that can simultaneously address the limitations of 2D cell culture and animal models. 16 For instance, patient-derived tumor organoids can well preserve the histopathological features and genetic background of the parental tumor, enable high-throughput drug screening, and require relatively short culture periods. 17 However, although these 3D models have shown potential in personalized drug screening, different types of culture methods significantly affect the culture effect and application field. For example, there are a variety of methods for culturing organoids, including submerged culture, hanging drop culture, bioreactor culture, and so on. Submerged culture is the most widely used method for culturing organoid. Cells or cell clusters are embedded in extracellular matrix (ECM) gels such as basement membrane extract or Matrigel and immersed in culture medium, which supplemented with growth factors. 18 In this method, oxygen must diffuse through the culture medium and matrix before reaching the core of the tissue, resulting in low diffusion efficiency and causing hypoxia in the central region. Furthermore, Matrigel still cannot fully mimic the true tumor stroma due to its highly dynamic and heterogeneous characteristics. More importantly, immune components are lacking in these models. Therefore, various culture systems that constructing immune microenvironment have been developed, such as organoid-immune cell coculture systems. In this coculture system, activated peripheral blood lymphocytes were mixed with organoids in a certain proportion for culture can be used to evaluate the sensitivity of patients to immunotherapy. 19 However, this organoid-immune cell coculture system constructed in a fixed proportion cannot truly simulate the individualized tumor immune microenvironment of patients. Therefore, there is an urgent need to establish novel preclinical models to address these challenges.
Recent studies have reported that an air–liquid interface (ALI) tumor model can preserve multiple tumor tissue components and be applied to evaluate drug responsiveness. 20 The tumor tissue is placed at the interface between supportive collagen gel matrix (below) and air (above). This configuration enables tumor to come into contact with air, simulating gas exchange. Meanwhile, nutrients are provided through the matrix infiltrating the culture medium, which helps maintain the complete 3D structure of the tissue and the interaction between cells. Therefore, compared with the above-mentioned models, this ALI system that directly culture the tumor itself is capable of preserving not only the integrity of the tumor structure but also its native immune microenvironment. This study applied this method to culture murine tumor fragments.
To systematically evaluate our platform’s capability in characterizing tumor microenvironments with distinct immune backgrounds, we intentionally selected the “immune-hot tumor” with high immune infiltration (colorectal cancer MC38) and the “immune-cold tumor” with poor immune infiltration (breast cancer 4T1). We hypothesized that our ALI platform can maintain their differences in the immune microenvironment and therapeutic responses to immunotherapy, thereby validating the relevance and application potential of this platform in tumor immunotherapy. We prepared tumor tissue fragments from 4T1 and MC38 mouse tumors and cultured them for 7–14 days. Results demonstrated sustained tumor growth throughout the culture period. To explore the application of this mini-tumor culture model in drug response evaluation, we conducted drug sensitivity experiments with chemotherapy and immunotherapy agents. Findings revealed distinct responses to chemotherapeutic drugs and anti-PD-1/PD-L1 antibodies across tumor fragments from different sources. Collectively, we demonstrated the practical utility of this mini-tumor culture as an individualized efficacy evaluation platform.
Method
Cell culture
Murine mammary tumor cell lines (4T1) and murine colorectal cancer cell line (MC38) were purchased from Procell Life Science and Technology Co., Ltd. (Wuhan, China). Cells were cultured in RPMI 1640 (PM150110, Procell) and supplemented with 10% fetal bovine serum (164210, Procell) and 1% penicillin/streptomycin (PB180120, Procell) in a humidified incubator at 37°C and 5% CO2 atmosphere.
Materials
Cisplatin (D807330) and 5-fluorouracil (F6173) were purchased from Macklin (Shanghai, China). Gemcitabine (S1714) was purchased from Selleck (Shanghai, China). Resiquimod/R848 (R131891) was purchased from Aladdin (Shanghai, China). Purified anti-mouse CD279 (PD-1) (114101) and purified anti-mouse CD274 (B7-H1, PD-L1) (124301) were purchased from Biolegend (San Diego, United States). Recombinant Mouse IL-2 (CK24) was purchased from Novoprotein (Suzhou, China).
Animal study
A 6-week-old female BALB/C and C57BL/6 mice were purchased from Guangzhou Yancheng Biotechnology Co., Ltd. (Guangzhou, China). Mice were maintained at a temperature of 22 ± 2°C and relative humidity of 55 ± 10%, under a 12-h light/12-h dark cycle. All mice were acclimatized for 1 week upon arrival in a specific pathogen-free (SPF) condition prior to experiments. A 100 μL suspension of 4T1 cells (5 × 105) or MC38 cells (1 × 106) were subcutaneously inoculated into each mouse. Mice received intraperitoneal (i.p.) injections of 1 mg/kg resiquimod (R848) every other day for a total of five administrations starting on day 8 after tumor inoculation. Tumor tissues with a volume of approximately 500–1000 mm3 were dissected.
Mini-tumor culture
The collagen gel matrix was prepared on ice by mixing collagen type I (354236, Corning), 10 × DMEM/F12 (PM150312P, Procell) at a ratio of 9:1, and the pH was adjusted to neutral with NaOH (S2770, Sigma). This reconstituted collagen gel solution was kept on ice to prevent gel solidification until added to 0.4 μm polypropylene membrane cell inserts of 24-well plate (TCS021024, Biofil). 200 μL of reconstituted collagen gel solution was added to the insert and incubated at 37°C for 30 min to solidify. Tumor tissues with a volume of approximately 500–1000 mm3 were dissected and washed with phosphate buffered saline (PBS). Fresh tumor tissues were cut into 1–3 mm³ fragments, resuspended in reconstituted collagen gel solution, and 200 μL was added to precoated inserts. After gel solidification, the insert was placed into an outer tissue culture dish containing 500 μL of complete culture medium, which consisting of RPMI 1640 medium supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin and 10 ng/mL IL-2, ensuring that the liquid level in the culture dish did not exceed the tumor fragments layer. The dish was incubated in an incubator at 37°C with 5% CO2, ensuring the humidity was greater than 95% to reduce liquid evaporation. On the following day, the culture medium was replaced with drug-containing medium for the continued culture.
Cell viability examination
Cell viability was assessed by a modified MTT assay (ST316, Beyotime) by extending the MTT reaction time and the formazan dissolution time. At the endpoint of drug treatment experiments, the culture medium was replaced with medium containing 0.5 mg/mL MTT solution, followed by incubating the plate at 37°C for 5 h. Medium was then carefully removed, and dimethyl sulfoxide was added to each well to dissolve the formazan crystals. Plates were gently shaken and incubated overnight for 12 h. The absorbance values at 570 nm were measured using a microplate reader.
Histological analysis and immunofluorescence
The collected tumor tissues were fixed in formalin overnight. Prior to fixation, the embedded tumor fragments together with collagen gel matrix were carefully removed from the insert membrane using sterile forceps and transferred to a 2 mL tube. After PBS washing, formalin was added for fixation. Dehydration was performed using 70%, 80%, 95%, and 100% ethanol, followed by clearing with xylene. Finally, the tissues were infiltrated and embedded in paraffin wax. Consecutive 4–5 μm sections were obtained from formalin-fixed paraffin-embedded blocks. After dewaxing, sections underwent histological analysis using hematoxylin and eosin (H&E) staining. For immunofluorescence analysis, deparaffinized sections were placed in a container filled with Tris-EDTA retrieval buffer and heated in a microwave oven for antigen retrieval (G1206-250ML, Servicebio). Subsequently, sections were permeabilized with 0.5% Triton X-100 in PBS for 20 min at room temperature. The Ki67 immunofluorescence analysis was carried out using a Ki-67 polyclonal antibody at a dilution of 1:800 (28074-1-AP, Proteintech). The CD45 immunofluorescence analysis was carried out using a CD45 polyclonal antibody at a dilution of 1:200 (20103-1-AP, Proteintech). The secondary antibody was used at a dilution of 1:500 (RGAR004, Proteintech). Nuclei were stained with DAPI (P0131, Beyotime). Images were captured at 20 × magnification using BioTek Cytation1 automated microscope (Agilent Technologies Inc.).
Statistical analysis
The data were expressed as means ± standard errors of the mean and analyzed by GraphPad Prism V10.0 (GraphPad Software, Inc., San Diego, CA). Statistical significance was evaluated with one-way analysis of variance (ANOVA) and Dunnett’s test. The value of statistical significance was set at p < 0.05.
Results
Establishment of the ex vivo mini-tumor culture
To establish an ex vivo mini-tumor culture (Fig. 1A), we subcutaneously inoculated 4T1 or MC38 cells into mice. Once tumors reached the desired volume, animals were euthanized and subcutaneous tumors were harvested. Tumors were minced into 1–3 mm³ fragments using sterile surgical scissors. These tumor tissue fragments were cultured ex vivo using an air-liquid interface (ALI) system to support tissue viability and structural integrity. To monitor the proliferation of tumor fragments, we performed a modified MTT assay by extending the incubation time of MTT to 5 h and the dissolution time of formazan to 12 h. As shown in Figure 1B–C, tumor fragments derived from both breast and colorectal tumors exhibited robust growth characteristics. The growth of tumor fragments was characterized over a period of 7 days. During the first 4 days of culture, 4T1 tumor fragments maintained stable cell number. Starting from day 5, the cell numbers of tumor fragments rapidly increased by over 150%, and this upward trend was sustained through day 7 (Fig. 1B). Similar growth pattern was also found in the culture of MC38 tumor fragments (Fig. 1C). To assess the long-term growth properties of tumor fragments in this culture system, we prolonged the culture period to 14 days. Both 4T1 and MC38 exhibited a gradual increase in cell numbers throughout the entire cultivation period (Fig. 1D–E).
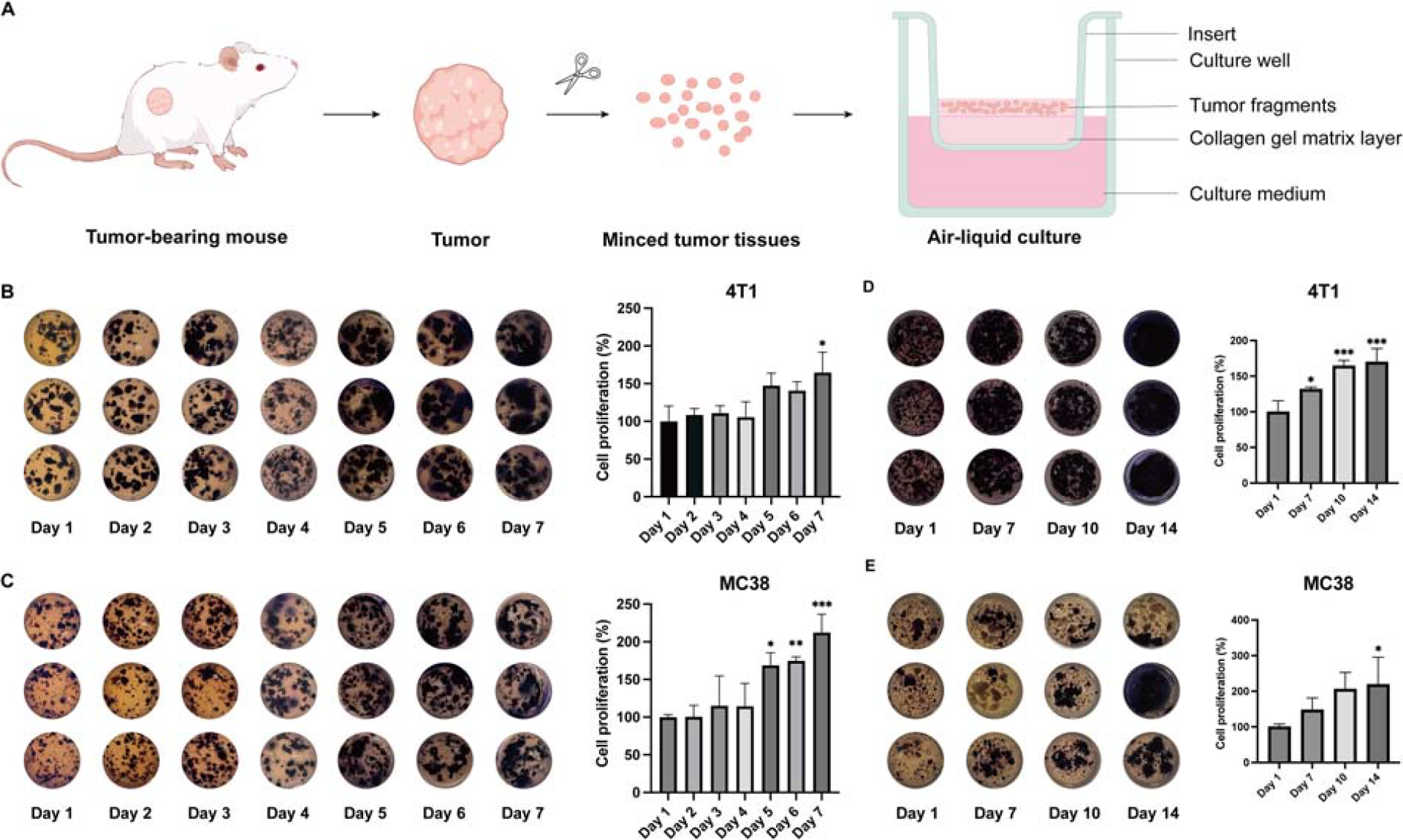
Establishment of the ex vivo culture model.
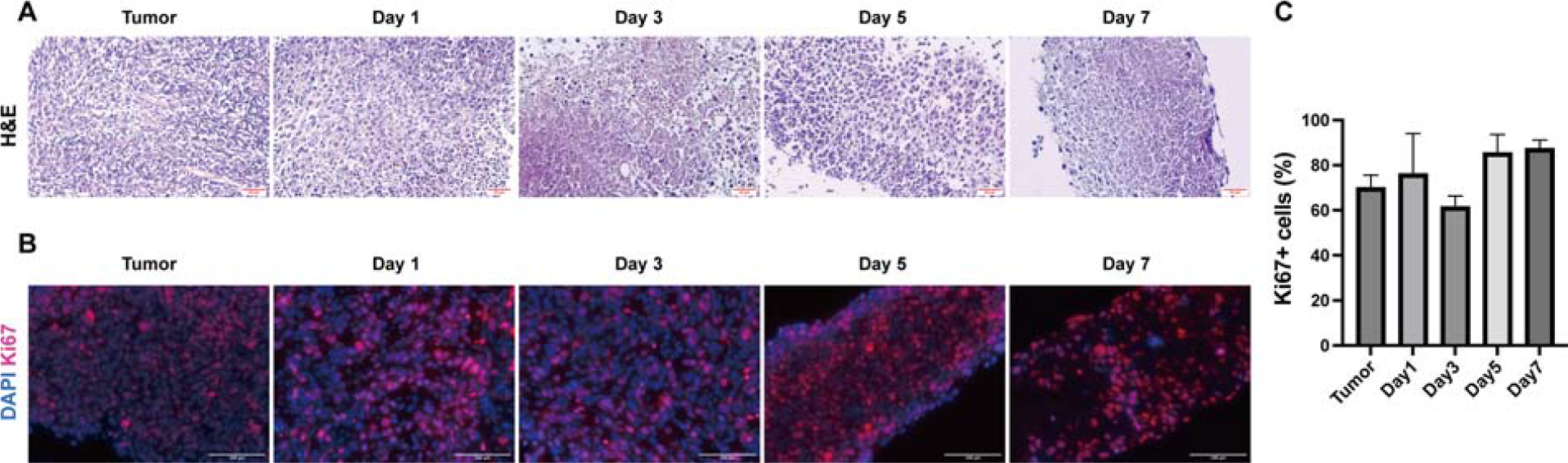
Subsequently, to further validate the robustness of mini-tumor culture model in sustaining tumor growth, we performed H&E staining on tumor fragments, which have been cultured for 0–7 days. Our results revealed that tumor fragments cultured for 1, 3, 5, and 7 days preserve gross histomorphology compared with the parental tumor tissues (Fig. 2A). This demonstrated that the mini-tumor culture model effectively preserved tissue morphology and cellular integrity for up to 7 days. Moreover, Ki67 staining provided further confirmation of the stable proliferation of the tumor fragments in this culture system. A consistently high level of Ki67 positive cells was maintained throughout the culture period indicating the sustained proliferative capacity (Fig. 2B–C). Thus, we have established an ex vivo mini-tumor culture platform capable of sustaining tumor growth for at least 2 weeks, providing a sufficient time window for drug sensitivity testing.
Ex vivo mini-tumor culture-based chemotherapy efficacy evaluation
The 4T1 and MC38 tumor fragments were treated with three chemotherapeutic agents for 6 days, including cisplatin, 5-fluorouracil and gemcitabine. Drugs were tested across various dose ranges, with cell viability evaluated by the MTT assay. As shown in Figure 3A, under cisplatin treatment, the viability of 4T1 and MC38 tumor fragments decreased in a dose-dependent manner, reducing from 77.21% at the lowest concentration (1.25 μM) to 37.25% at the highest concentration (20 μM), indicating their susceptibility to cisplatin. As shown in Figure 3B, 4T1 tumor fragments treated with 5-fluorouracil also exhibited a dose-dependent decrease in cell viability, declining from 82.27% at 1.25 μM to 52.04% at 20 μM. However, 5-fluorouracil did not cause a significant dose-dependent decrease in cell viability in MC38 tumor fragments. As shown in Figure 3C, we observed that the cytotoxicity induced by gemcitabine in both 4T1 and MC38 tumor fragments did not elevate with increasing concentration of gemcitabine, with the viability sustaining at approximately 50% across all tested gemcitabine concentrations. This phenomenon indicated that low doses of gemcitabine can effectively inhibit tumor growth but are insufficient for complete tumor cell clearance, suggesting gemcitabine may be more suitable for use in combination therapy. Collectively, these data demonstrated that our mini-tumor culture system can be used to evaluate chemotherapy efficacy.

Chemotherapy efficacy assessment using ex vivo mini-tumor culture model. 4T1 and MC38 tumor fragments treated with cisplatin
Ex vivo mini-tumor culture-based immunotherapy efficacy evaluation
To explore the application of mini-tumor culture model in evaluating immunotherapy responses, we examined the response of tumor fragments derived from “immune-cold” (4T1) and “immune-hot” (MC38) tumors to PD-1/PD-L1 checkpoint blockade. First, in order to confirm whether the tumor immune microenvironment characteristics of MC38 and 4T1 tumors are consistent with the existing literature, we conducted immunofluorescence examination on CD45+ immune cells in 4T1 and MC38 tumors. As shown in Figure 4A, there is a significant infiltration of CD45+ immune cells in the MC38 tumor, while this phenomenon is seldom observed in the 4T1 tumor. Furthermore, we also performed immunofluorescence staining for CD45 to mark infiltrated immune cells in the MC38 tumor fragments that cultured for 1 day and 7 days in the ALI system (Fig. 4A). The results showed that the CD45+ immune cell population were well maintained in the cultured tumor fragments throughout the 7-day culture period, without an obvious reduction in the proportion of immune cells. Next, the 4T1 and MC38 tumor fragments were treated with αPD-1 and αPD-L1 for 7 days. We observed that 4T1 tumor fragments didn’t response to both PD-1 and PD-L1 antibodies with cell viability over 85% (Fig. 4B). In contrast, for immune-hot MC38 tumor fragments, 10 μg/mL αPD-1 and αPD-L1 exhibited remarkably cytotoxicity decreasing tumor cell viability to approximately 60% (Fig. 4C). These findings suggested that our mini-tumor culture model can reflect the response of immune-cold and immune-hot tumor types to immunotherapy of PD-1/PD-L1 blockade.

Immunotherapy efficacy assessment using ex vivo mini-tumor culture model.
Immunologic adjuvants are believed to transform an immune-cold tumor microenvironment (TME) into an immune-hot TME by promoting immune cell infiltration. 21 We therefore chose the TLR7/8 agonist resiquimod (R848) to stimulate tumor reprogramming from an immune-cold to immune-hot stage. We investigated the therapeutic response of tumor fragments of R848-stimulated mice to PD-1 and PD-L1 antibodies. For the immune-cold 4T1 tumor fractions, compared with the tumor that did not receive R848 treatment (Fig. 4B), the immune responses targeting αPD-1 and αPD-L1 was significantly enhanced in the R848-stimulated tumor (Fig. 4D). The αPD-1 inhibition rate increased to 21.08%, and the αPD-L1 inhibition rate increased to 33.03% (Fig. 4D). Our data suggested R848 stimulation was able to convert immunotherapy-resistant cold tumors into responsive ones. For the immune-hot MC38 tumor, R848 treatment further increased the therapeutic efficacy of tumor fragments against αPD-1 and αPD-L1. The αPD-1 inhibition rate increased to 78.71%, and the αPD-L1 inhibition rate rose to 75.49% (Fig. 4C,E). Collectively, the mini-tumor culture model can reveal the immunotherapy responses of tumors with different immune types, providing robust experimental evidence for its potential utility in predicting immunotherapy efficacy.
Discussion
In this study, we developed a rapid and low-cost ex vivo mini-tumor culture protocol that successfully supported tumor growth and viability. This platform can be utilized for individualized drug response testing on anticancer chemotherapy and immunotherapy. Compared to other preclinical models, such as animal models, the ex vivo mini-tumor culture model offers the following advantages: (1) it significantly shortens the experimental cycle, enabling drug efficacy evaluation within 2 weeks; (2) it reduces the cost of experiment, as animal models often require substantial expenditures for animal procurement and maintenance, particularly highly immunodeficient transgenic mice; (3) it can be constructed from various types of solid tumor and used for high-throughput drug screening.
We generated tumor fragments from 4T1 and MC38 tumors and cultured them in vitro. Our results demonstrated that the mini-tumor culture model supported the proliferation of 4T1 and MC38 tumors within 2 weeks. Subsequently, we tested the efficacy of several representative chemotherapeutic agents on this model, including gemcitabine, 5-fluorouracil, and cisplatin. We observed differential therapeutic responses to these chemotherapeutic agents between 4T1 and MC38 tumors. For instance, at the same concentration (1.25 μM), gemcitabine caused a greater reduction in 4T1 tumor cell viability than cisplatin and 5-fluorouracil, indicating that 4T1 was the most sensitive to gemcitabine, followed by 5-fluorouracil and cisplatin. This finding is supported by published research that the IC50 values of gemcitabine in 4T1 cells was 15.05 nM after 24-h treatment, while the IC50 values of 5-fluorouracil and cisplatin were 8.9 μM and 14.2 μM after 48-h treatment, respectively.22,23
The tumor immune microenvironment is a highly structured ecosystem consisting of immune cells such as T cells, B cells, macrophages, natural killer cells and so on, as well as noncellular components such as immune-related cytokines/chemokines, molecules and ECM. TME plays a crucial role in tumor growth, angiogenesis, metastasis, drug response and resistance.24,25 Within the tumor immune microenvironment, the enrichment of regulatory T cells and myeloid-derived suppressor cells, coupled with the upregulation of immune checkpoint molecules such as PD-L1/CTLA-4, jointly construct a highly immunosuppressive ecosystem, promoting tumor immune escape and therapeutic resistance. 26 Cancer immunotherapy is a therapeutic approach that activates, enhances or reprograms the patient’s own immune system to specifically recognize, attack, and eliminate tumor cells. Currently, immunotherapy has cured some advanced or refractory cancers. However, most low-immunogenic tumors still exhibit low response rates to existing immunotherapies. Therefore, it is necessary to employ a preclinical pharmacodynamic platform to evaluate candidate immunotherapies.
Generally, tumors are categorized into “immune-cold” and “immune-hot” tumors based on the spatial distribution of immune cells within the tumor microenvironment. 27 Immune-hot tumors are characterized by extensive T cell infiltration both within and around the tumor, making tumors more responsive to immunotherapy. In contrast, immune-cold tumors exhibit less tumor infiltration of T cells and demonstrate relatively weaker responses to immunotherapy. 28 Therefore, we established mini-tumor culture model based on 4T1 and MC38 tumors and evaluated the therapeutic effects of αPD-1 and αPD-L1. We first profiled the immune cell infiltration in 4T1 and MC38 tumors. CD45+ immune cells were detectable in both tumor types, but were significantly more abundant in MC38 tumors. In addition, we also examined the maintenance of CD45+ immune cells in the ALI system and found that most of the CD45+ immune cells were well retained over 7-day culture period. Nevertheless, a partial loss of immune cells might occur after longer culture time, thereby affecting the assessment of immunotherapy efficacy. In general, we define our ALI mini-tumor culture platform as a short-term (7-day) ex vivo model for studying tumor-iimmuneinteractions and assessing immunotherapy responses. Through this mini-tumor culture model, the differential immunotherapy responses between immune-cold and immune-cold tumors were well demonstrated. Specifically, the MC38 tumor showed a higher sensitivity to the PD-1/PD-L1 blockade therapy than the 4T1 tumor. In fact, 4T1 is classified as immune-cold tumor, whereas MC38 is considered as immune-hot tumor.29–31
Previous research has demonstrated that immunologic adjuvants can transform an immune-cold TME into an immune-hot TME by promoting immune cell infiltration. 21 Our result corroborated this opinion that tumor immune infiltration is significantly increased in mice treated with resiquimod (R848), a new adjuvant targeting TLR7/8, which explains why such immune-cold 4T1 tumor respond to αPD-1/αPD-L1-based immunotherapy (Fig. 4D). Consequently, we demonstrated the mini-tumor culture model can reveal the immunotherapy responses of tumors with different immune types, providing robust experimental evidence for this model’s potential in predicting immunotherapy efficacy.
Tumor heterogeneity is a common characteristic of cancer, leading to varying responses among patients to different treatment regimens. Rarely does a single therapy prove suitable for all patients. Therefore, developing individualized treatment strategy is of paramount importance in cancer therapy. Individualized medicine, also known as precision medicine or personalized medicine, refers to a treatment approach that provides individual patients with the most suitable medication. The challenge in achieving individualized precision medicine lies in the fact that current gene sequencing-guided targeted drug strategies are only applicable to a small subset of patients. The majority of cancer patients cannot be detected with druggable mutations matching targeted drugs. Furthermore, a series of preclinical drug evaluation models face insurmountable difficulties in terms of timeliness and drug coverage. For instance, drug screening using PDX models requires an extended cycle (typically 3–6 months), failing to provide immediate guidance for clinical treatment. While organoid models are suitable for screening chemotherapeutic and targeted drugs, their limited simulation of the immune microenvironment renders their efficacy evaluation for immunotherapy drugs less reliable. Therefore, developing a new individualized therapeutic screening platform that enables rapid drug screening for immediate clinical guidance while highly simulating the tumor immune microenvironment to accurately evaluate immunotherapy efficacy is critically important. This study has preliminarily established such a mini-tumor culture protocol capable of achieving this goal, enabling immediate and feasible screening of chemotherapy and immunotherapy drugs for different tumor types.
Conclusion
In summary, we have established an ex vivo mini-tumor culture platform capable of sustaining tumor growth for at least 2 weeks and maintaining immune cell infiltration for 1 week, providing a sufficient time window for drug sensitivity testing. Using this mini-tumor culture platform, we can evaluate the individualized therapeutic responses of different tumors to chemotherapy and immunotherapy, including gemcitabine, 5-fluorouracil, cisplatin, αPD-1, and αPD-L1. Furthermore, using this drug evaluation platform, we revealed distinct therapeutic responses to immunotherapy between immune-cold tumors and immune-hot tumors, and demonstrated the important role of the immunologic adjuvant resiquimod (R848) in enhancing immunotherapy efficacy. This mini-tumor culture protocol provides a feasible implementation approach for ex vivo personalized drug testing.
Authors’ Contributions
Conceptualization: J.B., X.W., and R.-B.D. Validation: Y.F. and Y.Z. Formal analysis: Y.F. and Y.Z. Data curation: Y.F., Y.Z., Z.C., C.Z., D.L., T.D., and J.H. Writing—original draft preparation: Y.F. and Y.Z. Writing—review and editing: X.Z., J.B., X.W., and R.-B.D. Project administration: J.B., X.W., and R.-B.D. Funding acquisition: R.-B.D. All authors have read and agreed to the published version of the article.
Ethical Statement
This study was assessed and approved by the Animal Welfare and Ethical Review Committee of Hainan University for animal research (Approval No. HNUAUCC-2024-00036) and was conducted by following the human and animal research guidelines of the Hainan University.
Footnotes
Acknowledgments
The authors thank members of the Wang and Ding laboratories for helpful advice and discussion.
Author Disclosure Statement
The authors declare no competing interests.
Funding Information
This work was supported by the Hainan Provincial Natural Science Foundation of China (No. 822QN300), Joint Program on Health Science & Technology Innovation of Hainan Province (No. WSJK2024MS240), Hainan Province Science and Technology Special Fund (ZDYF2025SHFZ022), the National Natural Science Foundation of China (No. 82203206, 32560887, and 82360504), the Hainan University Collaborative Innovation Center Research Fund (No. XTCX2022JKB01) and the Hainan “Nanhai New Star” Project (No. 202309009).