
Abstract
Background:
Idiopathic pulmonary fibrosis (IPF) is an incurable disease marked by irreversible fibrosis and declining respiratory function. Nintedanib inhalation powder is a novel dry powder inhalation formulation of nintedanib consisting of Technosphere® particles that deliver active drug directly to the lungs, aiming to reduce systemic effects associated with oral treatment. The objective of the present work was to assess whether nintedanib dry powder inhalation (DPI) has the potential to provide improved safety and thus effectiveness compared with that of currently available therapeutic options for IPF.
Methods:
Three preclinical studies evaluated pharmacokinetic and toxicity outcomes associated with nintedanib inhalation powder. Two 28-day repeat-dose studies (one in rats and one in dogs), and a 6-month toxicity study in dogs assessed pulmonary and histological changes after inhaled nintedanib therapy. A phase 1, first-in-human, randomized, double-blind, placebo-controlled study evaluated the safety, tolerability, and pharmacokinetics of single and multiple ascending doses of nintedanib DPI versus placebo in healthy adults.
Results:
In the 28-day repeat-dose studies and the 6-month study, animals survived to scheduled necropsy without significant adverse findings, including no gastrointestinal adverse findings, and most achieved target pulmonary-deposited nintedanib concentrations. The phase 1 study demonstrated dose-proportional increases in nintedanib plasma concentrations after single doses and rapid absorption of nintedanib, with maximum concentrations within a few minutes of first dose at levels consistent with deep lung deposition. Nintedanib inhalation powder was considered safe and well tolerated throughout the study duration.
Conclusion:
Inhaled administration of nintedanib via DPI may offer advantages over oral therapies in patients with IPF, potentially reducing adverse effects associated with systemic exposure. Results of the preclinical and phase 1 studies support continued assessment of this formulation in patients with IPF and other pulmonary fibrotic diseases.
Clinical Trial Registration Number:
NCT06532942.
Keywords
Introduction
Interstitial lung disease (ILD) is a heterogeneous group of disorders causing inflammation and potentially pulmonary fibrosis. 1 Idiopathic pulmonary fibrosis (IPF) is an ILD characterized by progressive fibrosis causing irreversible loss of lung function, which may result in respiratory failure and death.2–6 IPF symptoms, most commonly progressive cough, dyspnea, and inspiratory bibasilar crackles, may contribute to impaired quality of life.6,7
IPF prevalence is associated with increased age (mean age 65–70 years).6,8,9 An estimated 100,000 and 34,000 people are living with IPF in the United States and Japan, 10 respectively, and the prevalence has increased since 2000, with a higher incidence in men.9,11,12 IPF is often accompanied by comorbidities that make prompt diagnosis and epidemiological studies challenging. 13
Currently available treatments slow fibrosis progression and manage symptoms, but none reverses scarring or cures IPF.14,15 In 2014, two oral antifibrotic agents, pirfenidone and nintedanib, were approved for IPF treatment in the United States.3,16,17 Pirfenidone is a modified pyridine small molecule with antifibrotic, anti-inflammatory, and antioxidant properties. 3 Nintedanib, a potent tyrosine kinase inhibitor, inhibits the effects of growth factors implicated in IPF pathogenesis.3,18 Although pirfenidone and nintedanib can slow IPF progression,19–21 oral doses needed to achieve therapeutic concentrations in lungs are associated with poor tolerability, particularly owing to gastrointestinal (GI) effects (eg, diarrhea) that may require dose modifications.19,21–23 The use of oral nintedanib or pirfenidone requires close monitoring of liver enzymes.16,17 Furthermore, these treatments are underutilized, with ∼13% of patients with IPF in a representative cohort in the United States having received nintedanib (1453/10,996) or pirfenidone (1448/10,996) and ∼43% of those discontinuing treatment (mean treatment duration, 10 months). 24
Lung transplantation is the only potentially curative treatment demonstrated to increase life expectancy3,25,26; however, few patients are eligible.5,27 New therapies (eg, oral nerandomilast and treprostinil solution) are in development.28–30 Combination therapies are on the horizon for IPF management but are not yet feasible owing to overlapping and compounded safety and tolerability profiles.30,31
Technosphere® (MannKind Corporation, Danbury, CT) technology is a drug delivery system that has been successfully used to deliver inhaled insulin to patients with diabetes mellitus.32,33 Its powder carrier, fumaryl diketopiperazine (FDKP), has also been used to deliver treprostinil via oral dry powder inhalation (DPI) for the treatment of pulmonary arterial hypertension (PAH) and pulmonary hypertension (PH) associated with ILD, inclusive of IPF. 34 Applying Technosphere technology to nintedanib would deliver therapeutic concentrations to lung tissues and may limit systemic exposure versus oral therapy. Overall, the advantages of inhaled therapy could translate to improved treatment persistence, effectiveness, and long-term outcomes.
Preclinical and phase 1 studies were conducted to evaluate the safety and pharmacokinetics (PK) of nintedanib inhalation powder and to assess the feasibility of further clinical investigation in people with IPF.
Materials and Methods
Nintedanib inhalation powder
Nintedanib inhalation powder consists of nintedanib-containing Technosphere particles composed of FDKP and polysorbate 80, both of which have been studied extensively in preclinical studies and humans in different programs, including two commercially available products.35–37 It is supplied in single-dose cartridges containing 10 mg of powder at 20% strength (ie, 2 mg nintedanib + 8 mg FDKP per cartridge) and administered via a proprietary inhaler. Cartridges can be stored at room temperature (20°C–25°C) outside the inhaler.
Preclinical studies
Inhalation toxicity and reversibility of nintedanib inhalation powder were evaluated in multiple animal studies, including two 28-day repeat-dose studies with 14-day recovery (rats and dogs) and a 6-month chronic toxicity study (dogs). Male and female Sprague-Dawley rats (N = 80) were randomized to low (0.5 mg/kg)-, mid (1.3 mg/kg)-, or high (2.3 mg/kg)-target pulmonary-deposited doses of nintedanib inhalation powder or air control (0 mg/kg) via nose-only inhalation once daily for 28 days, followed by a 14-day recovery. Toxicokinetic (TK) parameters, including total plasma and lung exposure (area under the concentration–time curve [AUC] from time zero to time of last quantifiable concentration [AUClast]), maximum plasma concentration (Cmax), and terminal elimination half-life (t1/2), were evaluated using blood and lung samples. Time points assessing TK parameters can be found in Supplementary Table S1.
In the second repeat-dose study, male and female beagle dogs (N = 42) were randomized to low (0.15 mg/kg)-, mid (0.4 mg/kg)-, or high (0.8 mg/kg)-target pulmonary-deposited doses of nintedanib inhalation powder or air (control) via face-mask inhalation once daily for 28 days, followed by 14 days of recovery. Time points assessing TK parameters are listed in Supplementary Table S1. In both the repeat-dose studies, mean achieved pulmonary-deposited doses were calculated for males and females within each treatment group and averaged between sexes.
In a 6-month toxicity study, beagle dogs (N = 32) were randomized to four groups (four male and four female dogs per group): filtered air control or nintedanib inhalation powder at nominal low (0.15 mg/kg)-, mid (0.4 mg/kg)-, or high (0.8 mg/kg)-target pulmonary-deposited daily doses for 180 days via face-mask inhalation. Blood was collected after exposures on days 1 and 180 (Supplementary Table S1) and predose on day 180 for TK (Cmax, AUClast). Lungs were assayed at sacrifice (24 hours after final exposure). Mean achieved pulmonary-deposited doses of nintedanib and other clinical observations were analyzed for male and female rats within treatment groups and averaged between sexes.
The studies were conducted in compliance with the US Food and Drug Administration 21 Code of Federal Regulations Part 58 (Good Laboratory Practices for Nonclinical Laboratory Studies) in Lovelace Biomedical’s animal research facilities, which are fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. These studies were conducted in accordance with all applicable sections of the Final Rules of the Animal Welfare Act regulations, as well as the Guide for Care and Use of Laboratory Animals (2011).
Phase 1 clinical trial
A phase 1, first-in-human, randomized, double-blind, placebo-controlled, two-part study was conducted to evaluate the safety, tolerability, and PK of single (SAD) and multiple ascending doses (MAD) of nintedanib inhalation powder in healthy adults (ages ≥40 and ≤65 years) at a single site in the United States (NCT06532942). 38 Full eligibility criteria for the study can be found in the Supplementary Data. Participants were randomly assigned 3:1 to nintedanib inhalation powder or placebo via inhalation.
Part A (N = 24) assessed SAD of 2, 4, or 8 mg of nintedanib versus placebo administered in the morning via inhalation (Fig. 1). Two sentinel participants received doses of nintedanib (n = 1) or placebo (n = 1) first. Following review of favorable safety data 48 hours postdose, the remaining participants received treatment. Pulmonary function tests assessing forced expiratory volume in 1 second (FEV1) and forced vital capacity (FVC) using spirometry were monitored predose and at 5, 15, and 30 minutes, and at 1, 1.5, and 2 hours postdose. PK samples were collected predose and at 5, 10, 15, and 30 minutes, and at 1, 2, 4, 6, 8, 12, and 24 hours after morning inhalation. Participants remained at the clinical research unit (CRU) on day 1 (predose) and were discharged upon completion of 24-hour postdose assessments on day 2.
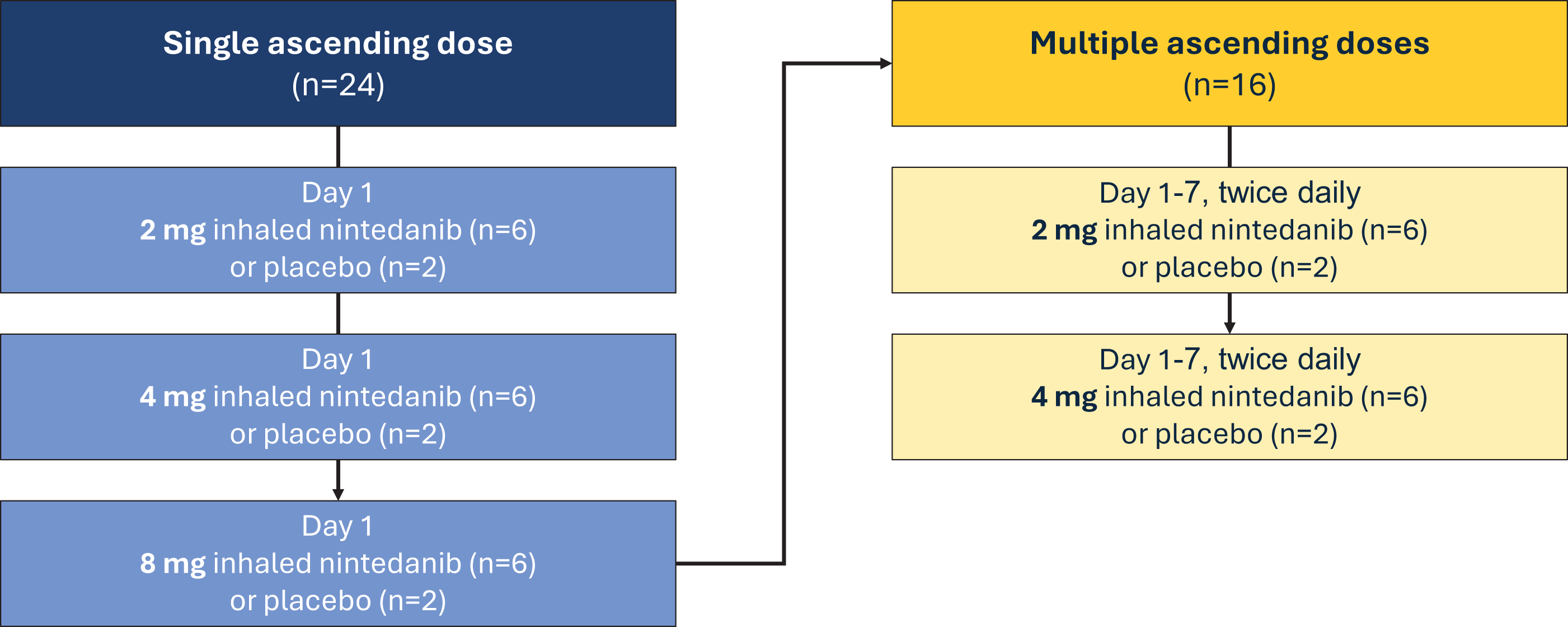
Single and multiple ascending dose phase 1 study design.
Following review of SAD safety data, Part B (MAD portion) randomized 16 additional participants to 2 or 4 mg nintedanib inhalation powder or placebo twice daily, ∼12 hours apart (Fig. 1). Pulmonary function tests were monitored on days 1, 4, and 7 predose and at 5, 15, and 30 minutes, and at 1, 1.5, and 2 hours postdose. PK samples were collected on days 1 and 7 predose and at 5, 10, 15, and 30 minutes, and at 1, 2, 4, 6, 8, and 12 hours after morning inhalation, and again 12 hours after evening inhalation. On days 2 through 6, PK samples were only collected before morning inhalation. Participants remained at the CRU for 8 nights and were discharged upon completion of the 12-hour day 7 dose assessments. Secondary PK outcomes based on plasma concentrations of nintedanib inhalation powder included Cmax, time to maximum concentration (Tmax), t1/2, AUC over a dosing interval (AUCtau), apparent total body clearance (CL/F), and apparent volume of distribution during the terminal phase (Vz/F).
Safety endpoints, treatment-emergent adverse events (TEAEs), and serious adverse events (SAEs) were summarized by cohort and treatment received using descriptive statistics. Additional safety endpoints can be found in the Supplementary Data.
No formal statistical hypothesis testing was conducted. Analyses were descriptive, providing summary statistics for quantitative data and frequency tables for qualitative/ordinal data. PK parameters were calculated for each individual and study day. PK parameters assessing inhaled nintedanib were estimated from plasma concentration–time data from each cohort following dose administration. Geometric mean and percent coefficient of variation were calculated for PK data.
Ethical conduct
The trial was conducted in accordance with the protocol, with ethical principles derived from the Declaration of Helsinki and Council of International Organizations of Medical Sciences International Ethical Guidelines, International Council for Harmonization Good Clinical Practice guidelines, and all applicable laws and regulations. The study protocol was reviewed and approved by the institutional review board: Advarra IRB (6100 Merriweather Dr., Suite #600, Columbia, MD 21044). All participants signed written informed consent forms before screening.
Results
Preclinical studies
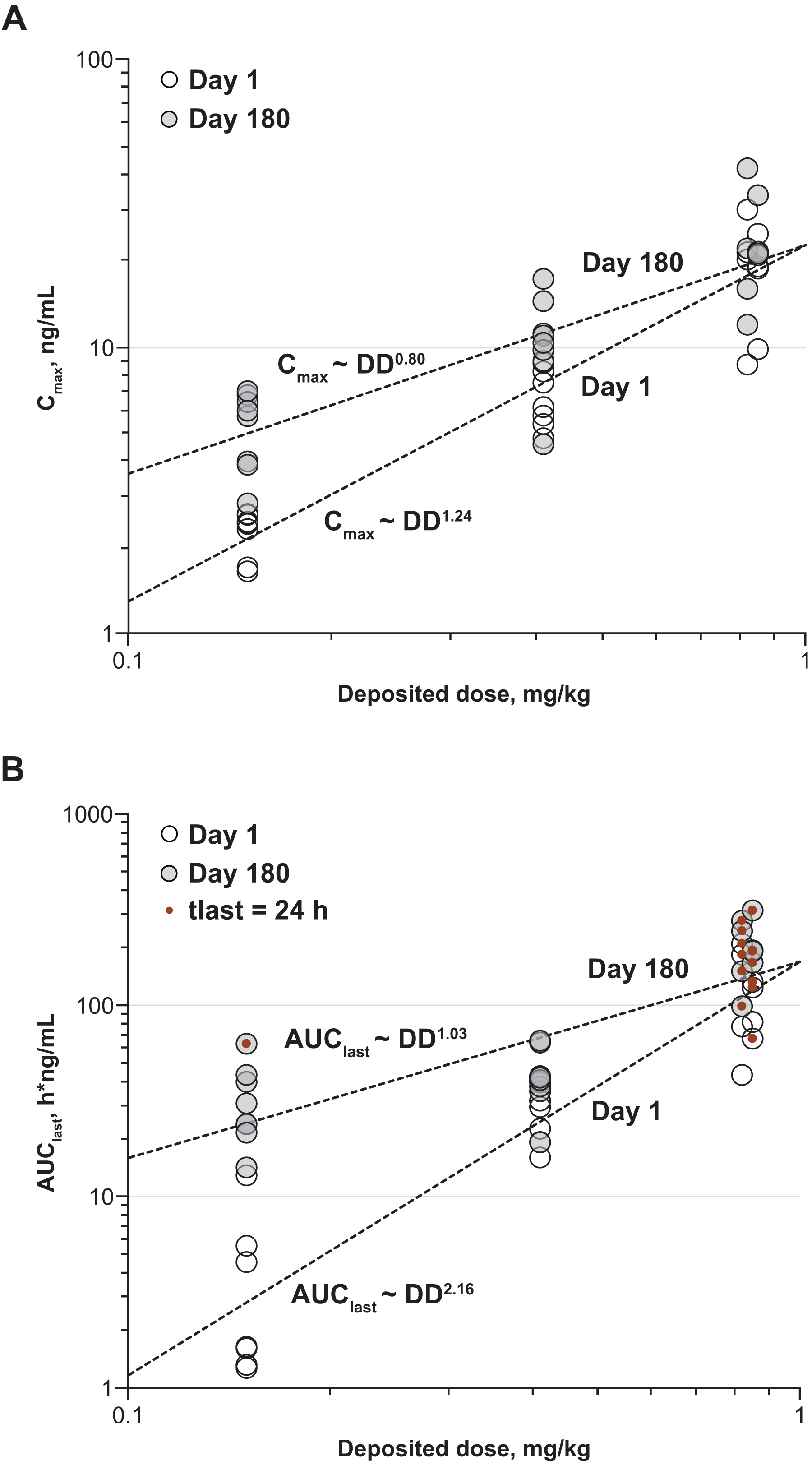
The repeat-dose study in rats showed no drug-related changes in laboratory assessments. Histological changes were limited to the respiratory tract at all examined doses and deemed nonadverse. Mean achieved pulmonary-deposited doses of nintedanib inhalation powder were 0.44, 1.38, and 2.64 mg/kg (Table 1). Greater than dose-proportional plasma concentrations and total plasma exposures were observed across the dose range for nintedanib inhalation powder on days 1 and 28 (Fig. 2A and B), and t1/2 after 28 consecutive doses ranged from 5 to 7 hours. Lung concentrations were ≥100-fold higher than plasma concentrations after a single dose on days 1 and 28 (Fig. 3A), and t1/2 ranged from 4 to 5 hours on day 1, and 6 to 8 hours on day 28. Total lung exposure for both sexes was generally dose proportional across the dose range (0.5 to 2.3 mg/kg target deposited dose, 0.43 to 2.71 mg/kg pulmonary-deposited dose) on days 1 and 28 (Fig. 3B). In the absence of adverse findings at any dose, the sex-averaged achieved high dose (2.64 mg/kg) was considered the no observed adverse effect level (NOAEL).
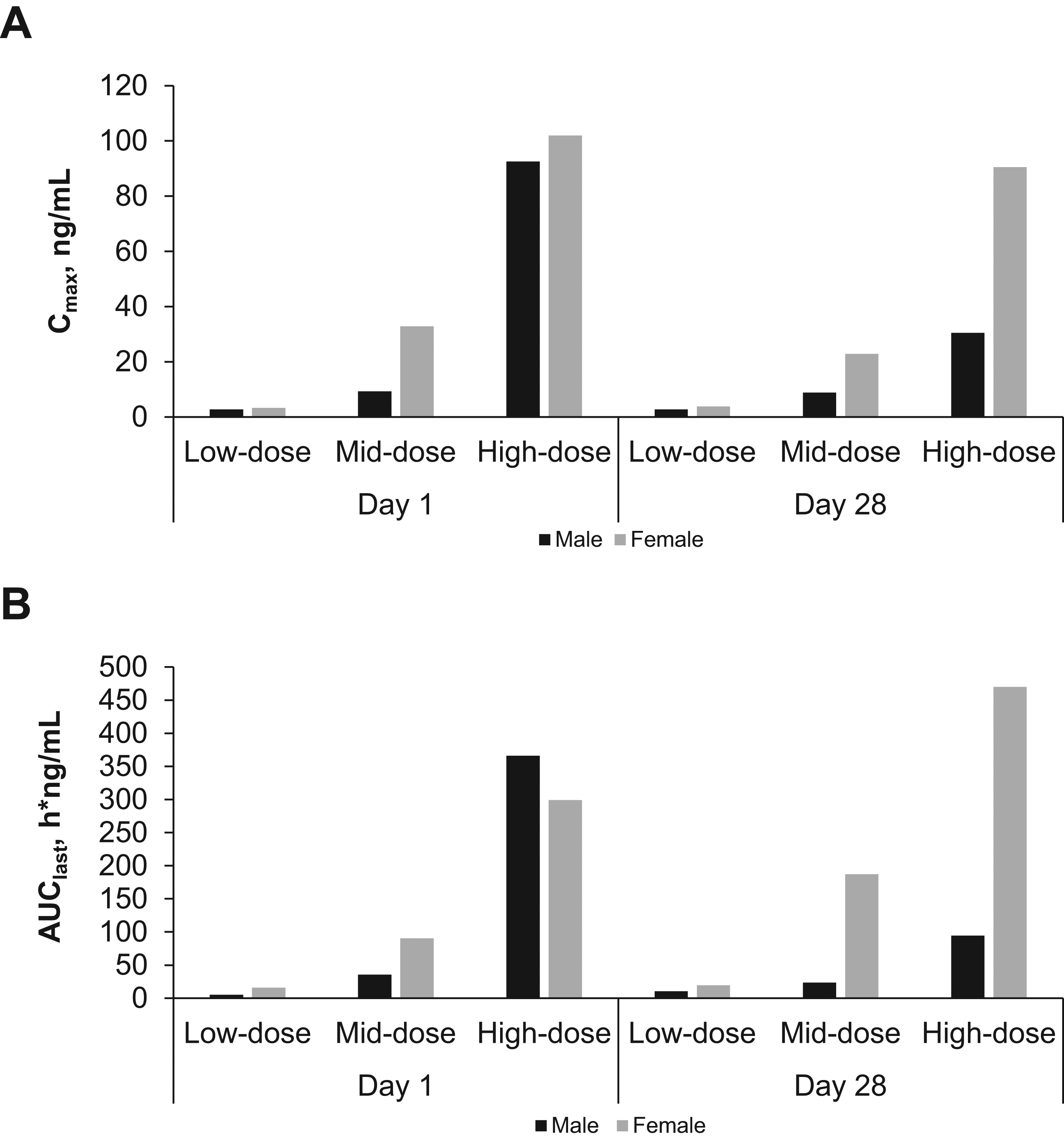
Mean plasma Cmax
Mean lung Cmax
Pulmonary-Deposited Doses of Nintedanib Inhalation Powder in 28-Day Repeat-Dose Rat and Dog Studies
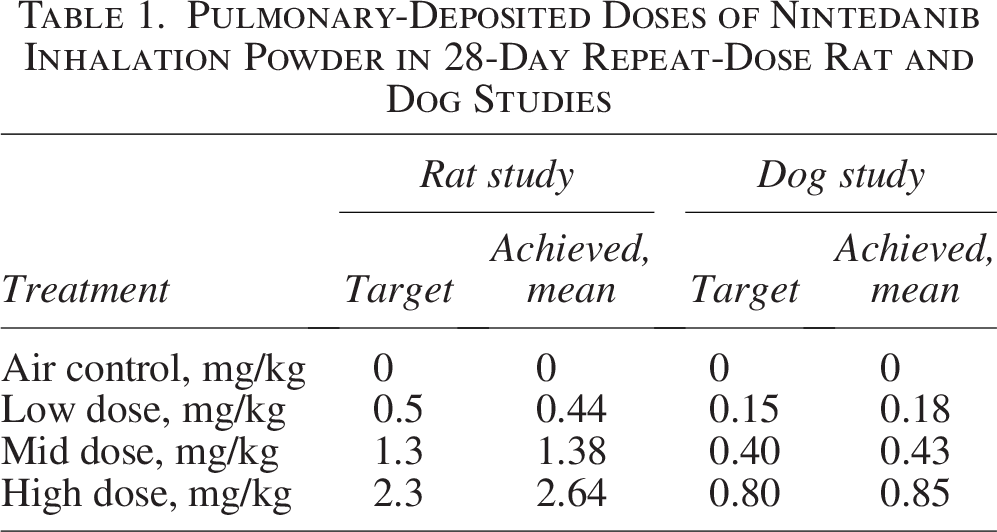
The repeat-dose study in dogs showed no associated clinical signs of toxicity throughout the study. Administration of nintedanib inhalation powder caused no perturbations in pulmonary function parameters. The achieved deposited doses of 0.18, 0.43, and 0.85 mg/kg were in line with their respective targets (0.15, 0.40, and 0.80 mg/kg), presented in Table 1. Exposure was dose related in plasma and lung tissue with measurable concentrations in lung samples 14 days after exposure in 75% (9/12) of the dosed recovery animals; nintedanib was below the limit of quantitation in all air control animals. Some inflammatory changes were present, but they were mild, did not affect all animals, and were deemed nonadverse. In the absence of adverse findings at any dose, the NOAEL was the sex-averaged high dose of 0.85 mg/kg.
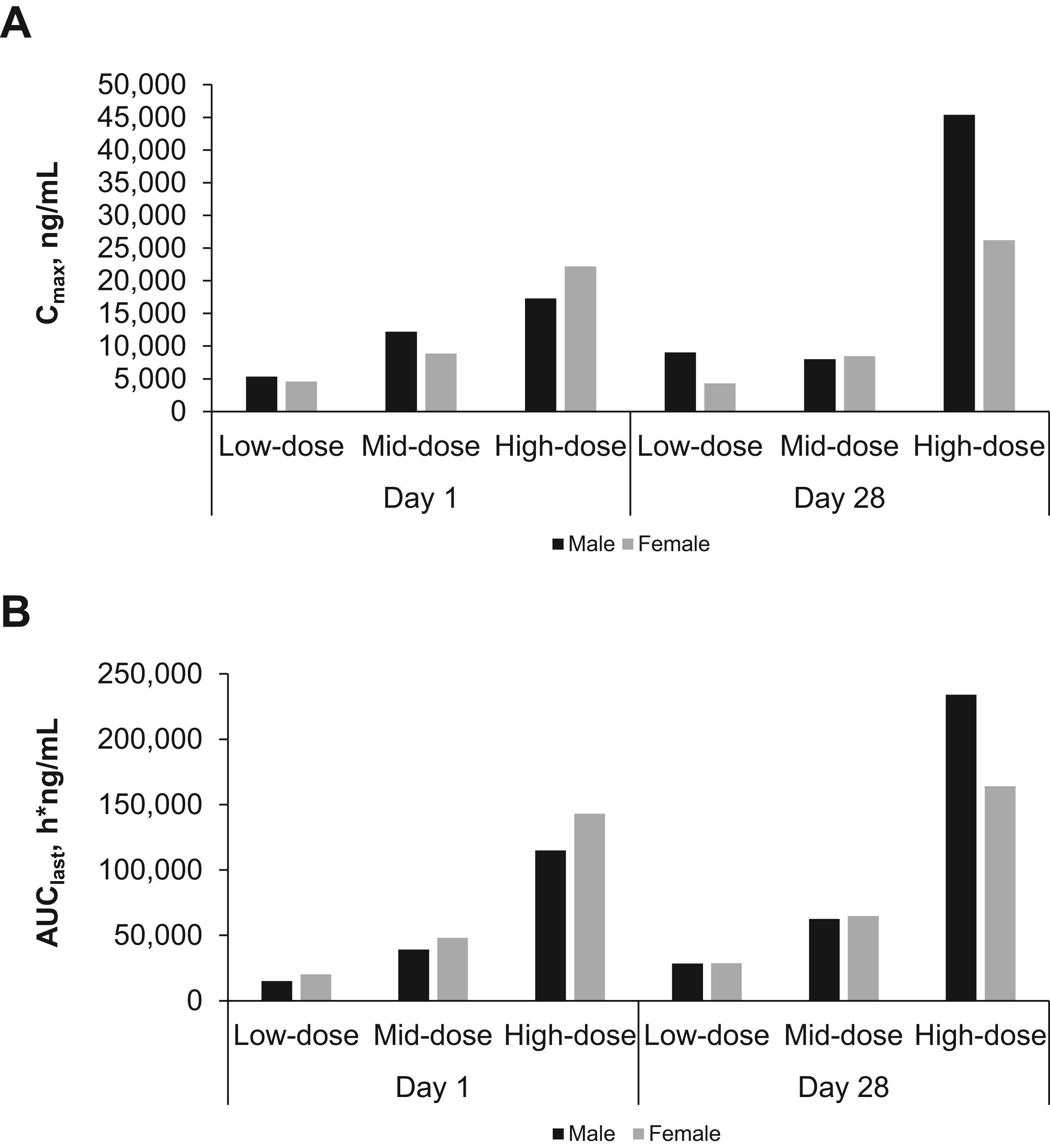
The 6-month toxicity study in dogs showed no associated adverse events (AEs). Cmax occurred at 20–40 minutes after exposure, and a clear dose-related response was evident in mean plasma profiles on days 1 and 180 (Fig. 4A and B). Actual sex-averaged pulmonary-deposited doses were 0.15, 0.41, and 0.84, consistent with their respective targets. Some concentrations in the low- and mid-dose groups were particularly low on day 1, resulting in super-proportional dose response on day 1 and weaker dependence on day 180 (Fig. 5A and B); this generated apparent dose dependence in accumulation ratios (day 180/day 1). Geometric mean accumulation ratio for treated animals was 1.62 for Cmax and 2.12 for AUClast.
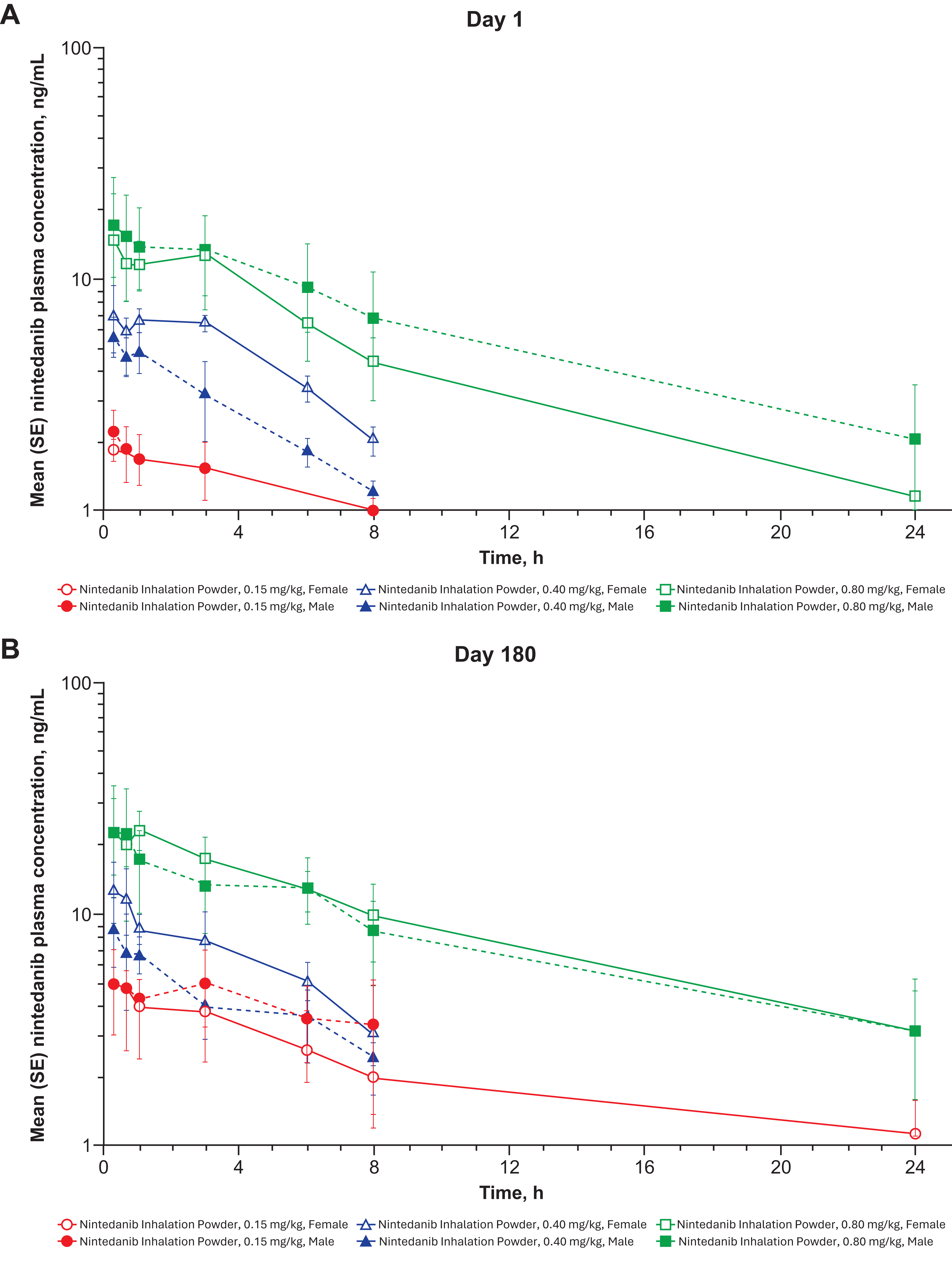
Mean plasma profiles from day 1
Plasma Cmax
Phase 1 study
A total of 40 participants (Part A, N = 18; Part B, N = 12; Placebo, N = 10) were enrolled, received nintedanib inhalation powder or placebo, and completed the study as per protocol. Participants were predominantly male (Part A, 12 [67%]; Part B, 7 [58%]; placebo, 8 [80%]) and White (Part A, 13 [72%]; Part B, 8 [67%]; placebo, 6 [60%]). Median age of the study population for Part A (N = 18) was 51 years (IQR, 43–57 years). Median age for Part B (N = 12) was 50 years (IQR, 45–58 years) (Supplementary Table S2).
Nintedanib plasma concentrations generally increased with dose after a single-dose administration of 2 to 8 mg nintedanib inhalation powder (Table 2). Variability in nintedanib plasma concentrations and calculated exposures was observed between participants on day 1 after both single (Fig. 6A and B)- and multiple (Fig. 7A and B)-dose administration and within participants based on multiple-dose administration on day 7 (Fig. 8A and B). Nintedanib was rapidly absorbed, with Tmax observed within a few minutes of DPI dose administration. Furthermore, t1/2 was similar between the dose groups after a single dose, with geometric mean values between 10.3 and 14.6 hours. For SAD, Cmax concentrations increased as dose increased; for MAD, Cmax increased for the 2 mg dose but not for the 4 mg dose. PK results from the MAD phase are shown in Table 2.
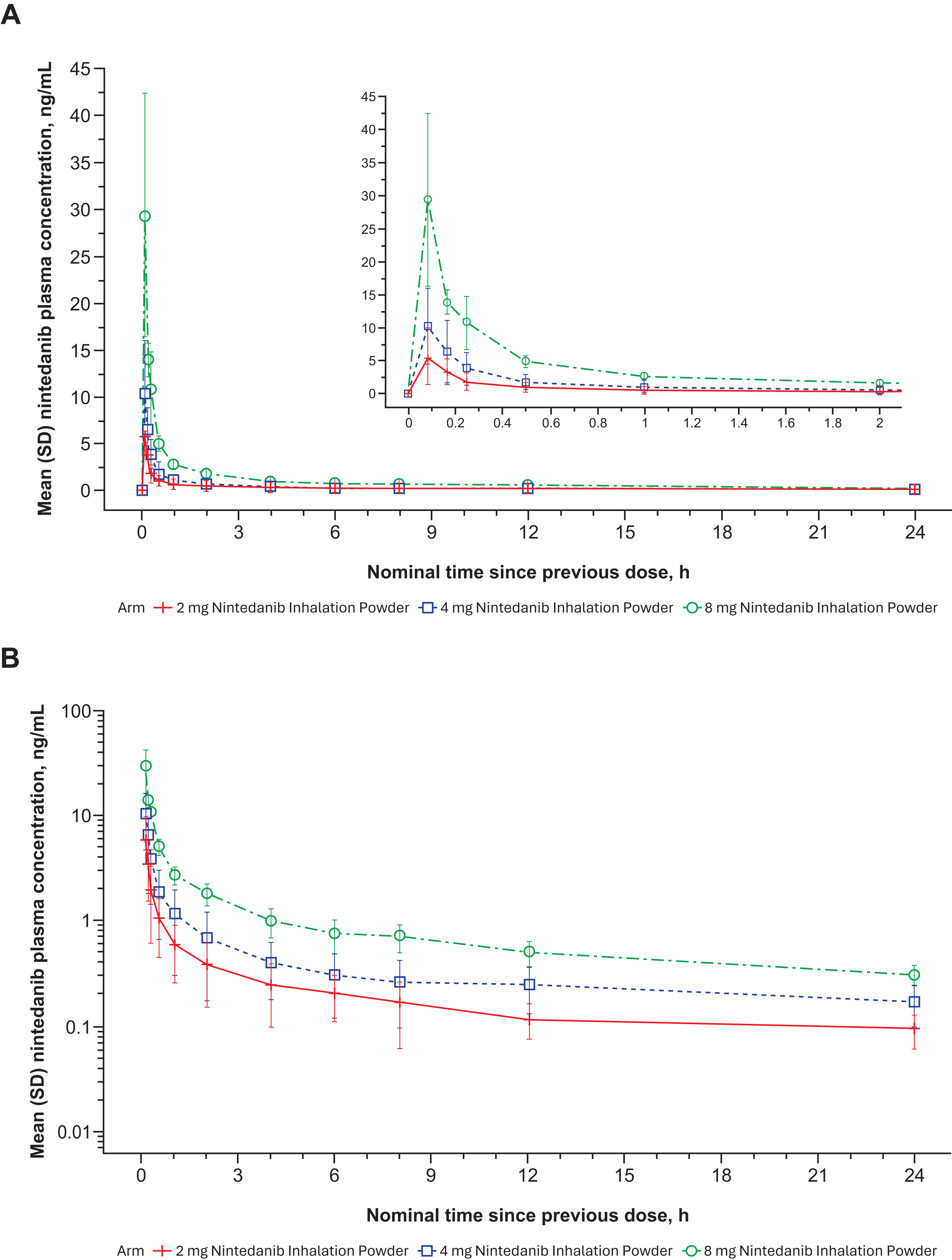
Mean nintedanib plasma concentration–time profiles after single ascending dose nintedanib inhalation powder from phase 1 study in healthy adult (>40 years old) volunteers (Part A) expressed both linearly
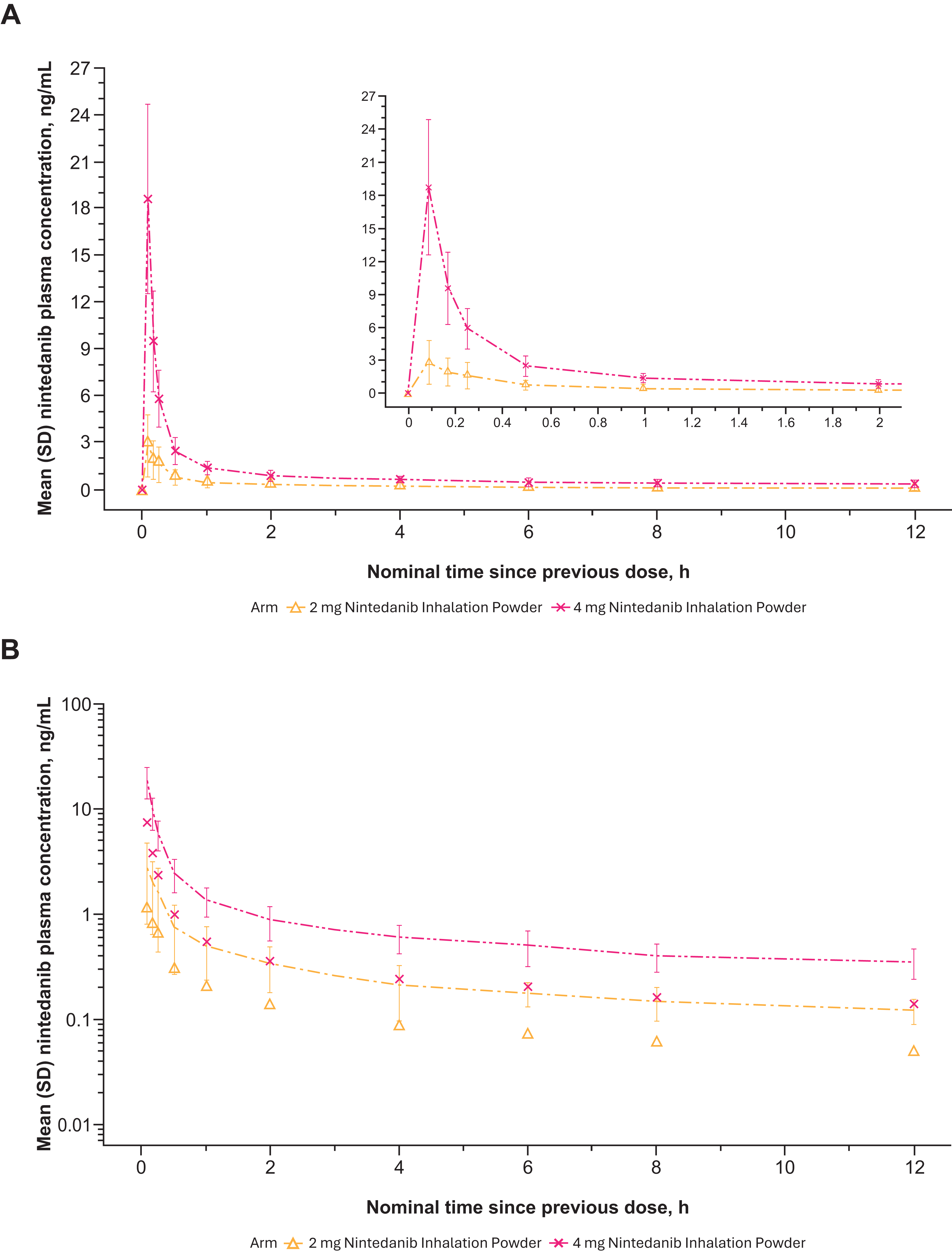
Mean nintedanib plasma concentration–time profiles after multiple ascending dose nintedanib inhalation powder on day 1 from phase 1 study in healthy adult (>40 years old) volunteers (Part B) expressed both linearly
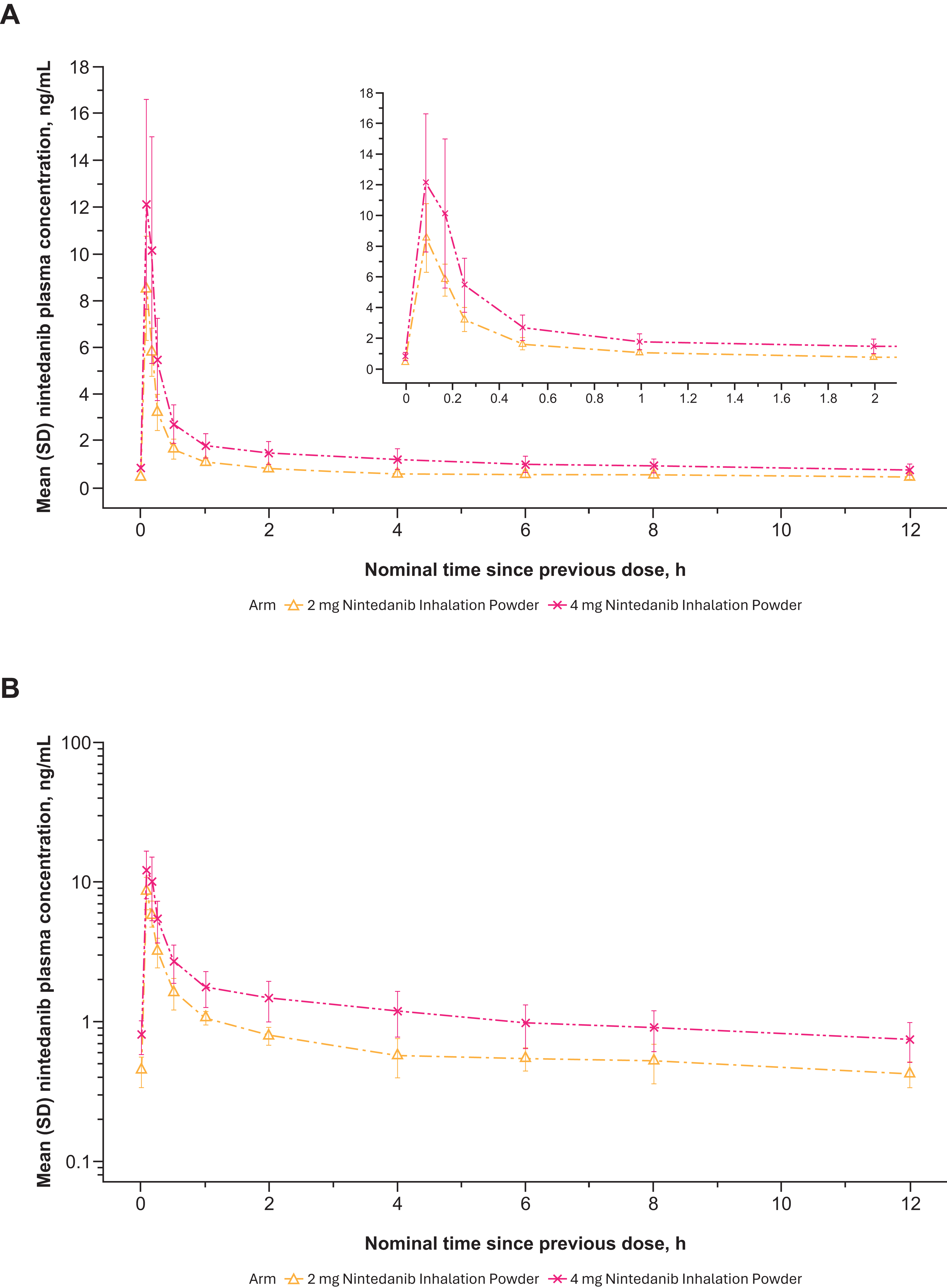
Mean nintedanib plasma concentration–time profiles after multiple ascending dose nintedanib inhalation powder on day 7 from phase 1 study in healthy adult (>40 years old) volunteers (Part B) expressed both linearly
Geometric Mean of Nintedanib Pharmacokinetic Parameters After Single Ascending Dose and Multiple Ascending Dose in Phase 1 Study of Healthy Adult (>40 Years Old) Volunteers
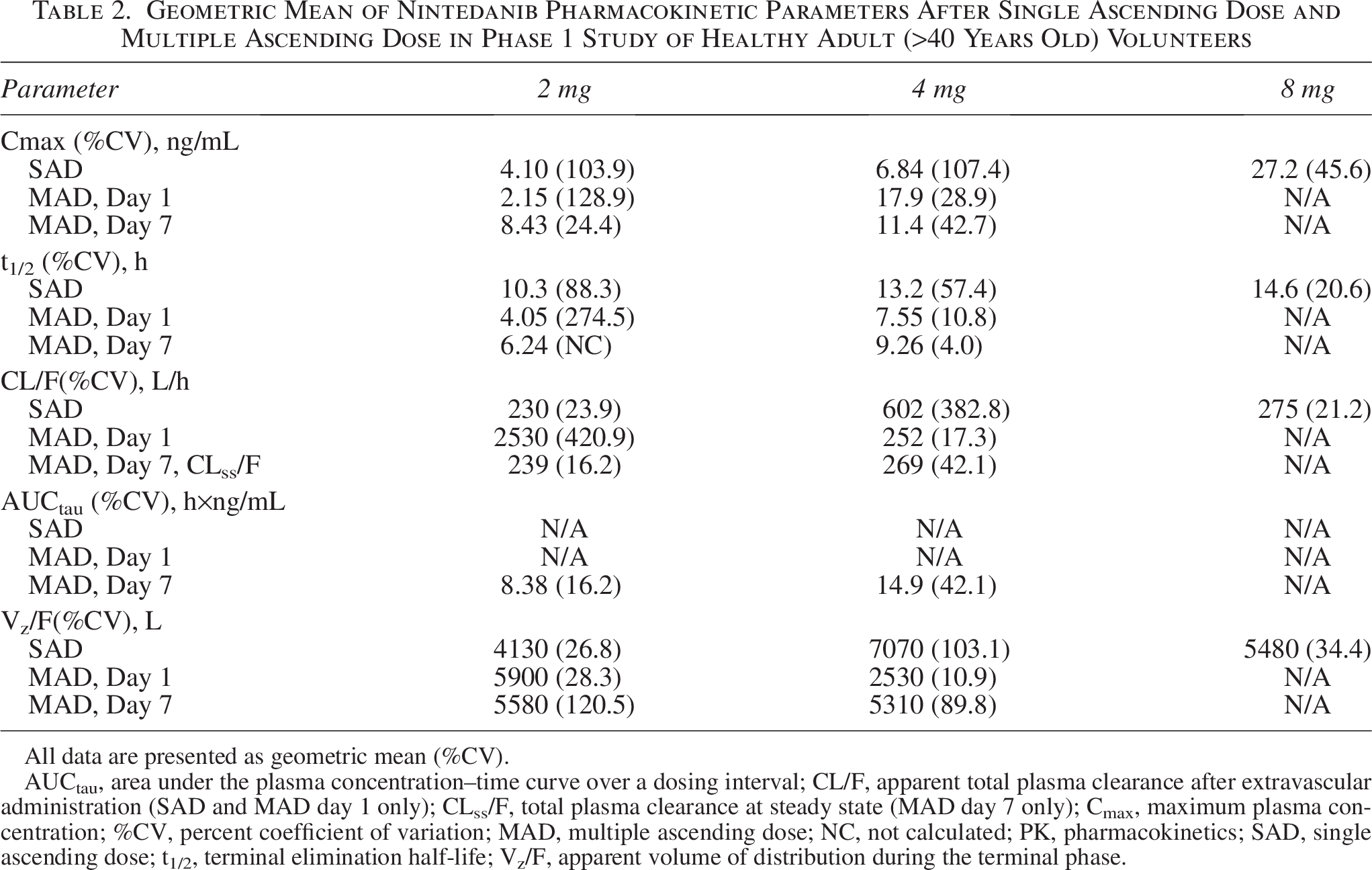
All data are presented as geometric mean (%CV).
AUCtau, area under the plasma concentration–time curve over a dosing interval; CL/F, apparent total plasma clearance after extravascular administration (SAD and MAD day 1 only); CLss/F, total plasma clearance at steady state (MAD day 7 only); Cmax, maximum plasma concentration; %CV, percent coefficient of variation; MAD, multiple ascending dose; NC, not calculated; PK, pharmacokinetics; SAD, single ascending dose; t1/2, terminal elimination half-life; Vz/F, apparent volume of distribution during the terminal phase.
All reported TEAEs were mild and resolved. No SAEs, severe TEAEs, TEAEs leading to discontinuation or dose adjustments, or deaths were reported (Table 3). Only cough and FEV1 reduction of ≥15% were reported as AEs. Of 21 total participants who had TEAEs, 18 experienced a cough. A ≥15% reduction in FEV1 from baseline and predose was reported as a TEAE in five participants within 30 minutes of the first dose of nintedanib inhalation powder. No treatment-related trends were observed, and events were transient and reversible within <1 hour in the active study arms. None of the AEs worsened during the study or led to study drug discontinuation.
Summary of Treatment-Emergent Adverse Events from Phase 1 Study in Healthy Adult (>40 Years Old) Volunteers
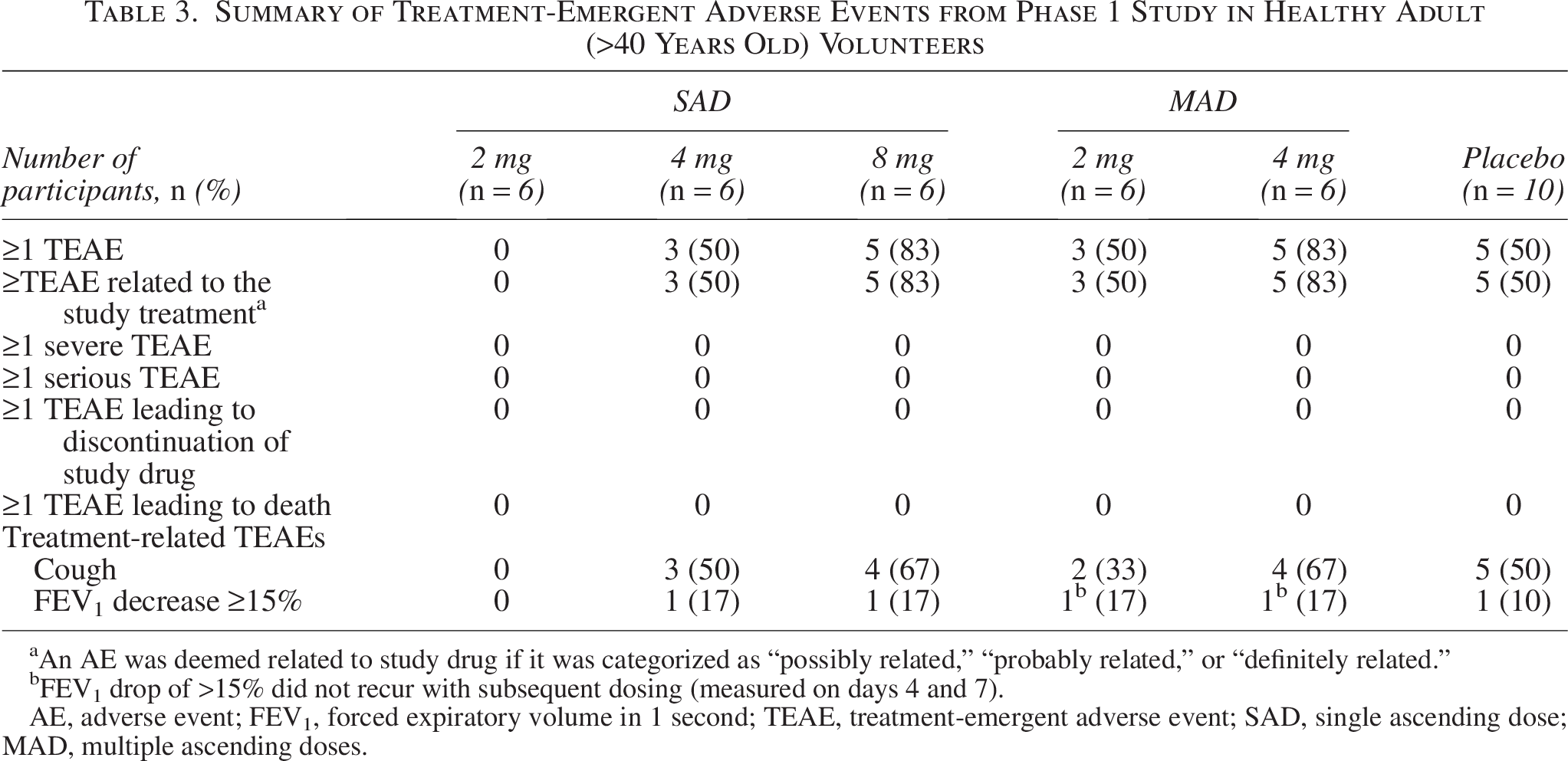
An AE was deemed related to study drug if it was categorized as “possibly related,” “probably related,” or “definitely related.”
FEV1 drop of >15% did not recur with subsequent dosing (measured on days 4 and 7).
AE, adverse event; FEV1, forced expiratory volume in 1 second; TEAE, treatment-emergent adverse event; SAD, single ascending dose; MAD, multiple ascending doses.
There was no dose response in FEV1 change or cough; cough was more common after the first dose, regardless of overall dose. Total mean FEV1 (L) change from baseline at 5 minutes and 2 hours postdose (day 1) was −0.24 and −0.15, respectively, during the SAD phase and −0.17 and −0.07 during MAD. Total mean FVC (L) change from baseline 5 minutes and 2 hours postdose (day 1) was −0.28 and −0.18, respectively, during the SAD phase and −0.20 and −0.15 during MAD. There was no meaningful difference in spirometry changes between active study arms and placebo, with significant spirometry variability in all groups. Spirometry changes were not associated with symptoms, and no participants had symptoms suggestive of bronchospasm. Asymptomatic spirometry changes did not worsen with ongoing use. There were no drops of >5% in spirometry parameters, for example, on day 4 of the study. There were no GI, neurological, or other systemic AEs.
Discussion
Preclinical studies in rats and dogs evaluated the TK and PK of nintedanib inhalation powder. Repeat-dose, 28-day studies assessed the toxicity and reversibility of nintedanib inhalation powder after 28 days of treatment followed by 14 days of recovery. Mean target pulmonary-deposited doses of drug were achieved in all treatment groups, except the low-dose group of the rat study. Results of the 6-month toxicity study in dogs were consistent with 28-day studies.
The phase 1 clinical trial in healthy adults demonstrated dose-proportional increases in nintedanib plasma concentrations after a single-dose administration. For multiple-dose administration, Cmax increased for the 2 mg dose but not for the 4 mg dose, likely owing to the small sample size and variability. Nintedanib was rapidly absorbed with maximum concentrations within a few minutes after the first inhaled dose, and nintedanib inhalation powder was considered safe and well tolerated throughout the study. Transient decreases in FEV1 and FVC were noted in both active and placebo arms, without associated symptoms (other than some with mild, dry, short-lasting cough) and no change in vital signs or oxygen levels. Some such changes can be expected with DPI, and Technosphere insulin is notably contraindicated in patients with chronic lung disease and carries an associated warning for acute bronchospasm. Treprostinil DPI also includes a warning for potential bronchospasm. The clinical significance of these changes, if any, in patients with pulmonary fibrotic diseases, is to be determined in subsequent clinical trials.
Bronchoalveolar lavage was not done as drug concentrations collected using this method are variable and reflect airway drug concentrations rather than interstitial lung concentrations. Plasma drug concentrations better reflect the extent and timing of deep lung drug deposition. IPF treatments may have negative systemic effects owing to high plasma concentrations, which results in a high rate of treatment discontinuation. 24 The safety and tolerability of nintedanib inhalation powder demonstrated in the phase 1 study lay the groundwork for further clinical studies in patients with IPF and other pulmonary fibrotic diseases. In particular, no SAEs were observed in any treatment groups; only mild TEAEs were reported. Patients with hepatic disease or abnormal liver function tests were excluded; however, further analysis is warranted to assess whether inhaled nintedanib reduces hepatotoxicity compared with oral formulation. It is reasonable to hypothesize that hepatotoxicity or other GI or neurological AEs of nintedanib inhalation powder will be extremely unlikely based on the very low systemic concentrations seen in this study and because an inhalation route bypasses the GI tract and the first-pass metabolism/higher liver exposure. 39 This sets nintedanib inhalation powder as a potentially better combination with oral pirfenidone, nerandomilast, and other future IPF therapies.
The rapid absorption and short Tmax observed for nintedanib inhalation powder in the current study also support further investigation. Nintedanib is a substrate of P-glycoprotein transporter, and the oral formulation may have limited absorption and bioavailability owing to transporter effects. 40 Although Cmax increased in both the 2 and 4 mg treatment groups during single-dose administration, increases were only observed in the 2 mg treatment group during multiple-dose administration. The decrease in Cmax from 17.9 ng/mL on day 1 to 11.4 ng/mL on day 7 is attributed to the small sample size and high variability of the 4 mg group. Additional data are needed to better understand changes in Cmax following multiple daily doses.
Oral nintedanib is already approved for the treatment of IPF. If nintedanib inhalation powder demonstrates slowed progression of IPF, it could open avenues toward treating other pulmonary fibrotic diseases, including in combination therapy. It uses an inhaler currently used in patients with pulmonary fibrosis for treatment of PH in the setting of ILD, inclusive of IPF. 34 Such DPI devices are convenient to use based on their compact size and high portability, compared with other inhaled delivery devices. 41
Nintedanib inhalation powder also utilizes the inert excipient FDKP, the same powder carrier used to deliver treprostinil via oral DPI for treatment of PAH and PH associated with ILD with Technosphere technology. 34 However, FDKP comprises only 80% of nintedanib inhalation powder compared with 99% of treprostinil DPI. 34 Following inhalation, FDKP is rapidly absorbed into the systemic circulation and is eliminated unchanged through renal excretion. 36 The safety and tolerability of FDKP are well established and characterized with two currently available DPI products, Technosphere insulin and treprostinil DPI.33,34,36
Treprostinil DPI is approved for the treatment of PAH and PH associated with ILD, including in patients with PH in the setting of pulmonary fibrosis including IPF. 34 In a 3-week study assessing treprostinil DPI in a patient population living with PAH, incidence of cough was 35%, with no SAEs considered related to study treatment.34,42 Incidence of cough decreased to 14% in the extension period among patients with a median (IQR) treatment duration of 107 (77, 140) weeks. 43 Results of a phase 1 study of nintedanib inhalation powder demonstrated similar results to what was seen with the use of treprostinil DPI; cough did not worsen with increased dose or cartridge intake. 43 However, no AEs involving cough led to drug discontinuation or necessary dose adjustment in the present study of nintedanib inhalation powder.
Conclusions
The safety and tolerability profile of nintedanib inhalation powder in healthy adults warrants further study in patients with pulmonary fibrotic diseases. These results lay the foundation for a phase 2 clinical study investigating the safety, tolerability, and efficacy in patients living with IPF and other pulmonary fibrotic diseases.
Authors’ Contributions
W.H.F.: Conceptualization, methodology, validation, formal analysis, writing—review and editing, and project administration. M.C.: Methodology, validation, formal analysis, and review and editing.
Footnotes
Author Disclosure Statement
All authors were employees and/or stockholders of MannKind at the time of research.
Funding Information
This study was funded by MannKind Corporation. Medical writing support was provided by Alison Thomas, PharmD, and Sanjula Wickramasinghe, PhD, from Citrus Health Group (Chicago, IL) and was funded by MannKind Corporation (Danbury, CT).
Institutional Review Board and Approval
Advarra IRB; IRB Registration Number: 00000971; Study Number: Pro00078691.
Supplemental Material
Abbreviations used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.