
Abstract
Background:
Inhalation of corticosteroids, bronchodilators, antivirals, and antibiotics is well established to treat a variety of pulmonary diseases; however, no inhaled receptor tyrosine kinase (RTK) inhibitors have so far been approved for clinical use despite the key role of RTKs in several pulmonary diseases. We describe a detailed roadmap to identify, optimize, and derisk an inhaled platelet-derived growth factor receptor (PDGFR) inhibitor with extended activity in the lungs.
Methods:
In this study, stable isotopes were used to model receptor turnover in vivo, a high-resolution crystal structure was generated to support structure-based drug design and enhancement of both potency and specificity, and critical physicochemical parameters that drive lung retention were identified. Since some RTK inhibitors are linked to adverse interstitial lung disease in humans, we developed a cell painting assay that helped to eliminate compounds with non-specific effects.
Results:
We describe the steps we took to optimize the conditions for nebulized delivery to the deep lung and confirmed that >90% of PDGFR inhibition was maintained for at least 6 hours after nebulization of a 1 mg/kg dose in rats. Finally, modeling was used to calculate the projected human dose for this molecule.
Conclusions:
While the focus of this article is on the identification of an inhaled PDGFR inhibitor, our approach to develop a highly potent inhaled compound that has extended lung retention to minimize systemic effects could be adopted for other RTK drug discovery or lung targeting approaches.
Introduction
Inhaled drugs, including corticosteroids, bronchodilators, antivirals, and antibiotics is used to treat a variety of diseases. In addition, an inhaled prostacyclin analog, iloprost, is approved to treat pulmonary arterial hypertension (PAH), and inhaled deoxyribonuclease improves lung function in Cystic Fibrosis patients. 1 However, until now, no inhaled receptor tyrosine kinase (RTKs) inhibitors have been approved for clinical use. Several RTKs play a role in pulmonary diseases2–4 and optimized inhaled RTK inhibitors could be used to specifically target these signaling pathway in the lungs while avoiding high systemic exposure levels that could result in adverse effects.
One example is the platelet-derived growth factor (PDGF) pathway. PDGFs were identified as platelet-derived mitogens that stimulated fibroblast and smooth muscle (SMC) proliferation.5,6 Once activated, PDGF receptors (PDGFR) phosphorylate downstream signaling cascades, resulting in cell growth and chemotaxis. 7 Treatment of patients with advanced PAH with the RTK inhibitor imatinib increased exercise capacity and decreased pulmonary vascular resistance. However, imatinib was poorly tolerated, with a 27% discontinuation rate versus 9% in the placebo group. In addition, subdural hematomas were observed in 8 patients that were on vitamin K antagonists for anticoagulation. 8 An inhaled PDGFR inhibitor with extended lung retention therefore could theoretically result in similar improvements in hemodynamics and exercise capacity while avoiding the systemic adverse effects that prevented approval of imatinib for PAH.
A key challenge for inhaled drug delivery is to maintain sufficient drug levels in the lungs while limiting systemic drug exposure, which is compounded by the high degree of vascularization in the lungs, thin alveolar and capillary membranes, and mucociliary clearance. 9 Lowering solubility or permeability, decreasing the off-rate, or improving the lipophilicity or basicity of a compound are several strategies that are described to improve lung retention. 10 There are, however, trade-offs that need to be made. For example, reducing permeability would also reduce inhibition of intracellular targets. In addition, highly potent compounds are required to minimize the inhaled dose.
Given the lack of clinically approved inhaled RTK inhibitors, we wanted to share our experience in developing an inhaled PDGFRβ inhibitor, JNJ-PDGFRi-1. We provided a detailed description of physicochemical properties that drive lung retention and generated a new PDGFRβ crystal structure that allowed structure-based design to improve potency while minimizing off-target activity. Our lead identification was further facilitated by using a cell painting assay to rule out potential deleterious effects early on during the screening phase. Extensive work was done to characterize the pH-dependent viscosity and solubility profiles of JNJ-PDGFRi-1 to maximize deep lung deposition. This article, therefore, describes several key steps to develop novel inhaled drugs and this strategy could be applied to identify drugs for a range of diseases, including PAH, chronic obstructive pulmonary disease (COPD), asthma, and idiopathic pulmonary fibrosis.
Materials and Methods
Detailed methods are available in the online Supplementary Methods, including for animal studies, kinase assays, kinase selectivity, in vivo PDGFRβ half-life, cascade impactor, and pharmacokinetic (PK) measurements.
Ethical approval statement
All protocols for animal studies were approved by the
Structure determination of JNJ-PDGFRi-1 bound to PDGFRβ
Purified PDGFRβ protein (residues 557-981) was used to generate crystals in VDXm hanging drop plates. Once crystals had grown, several were transferred to a soaking solution containing 5 mM JNJ-PDGFRi-1. Diffraction data were collected at the Advanced Photon Source IMCA-CAT 17ID beamline. Datasets were processed using autoPROC 11 and solved by molecular replacement using apo PDGFR-β as a search model in Phenix. 12 Figures were generated with PyMol (Schrödinger).
Cell painting
Cell painting was based on the literature, 13 with parameters optimized for rat A10 cells (American Type Culture Collection, ATCC). Cells were treated with 10 ng/mL PDGF-BB overnight and then stained with MitoTracker (ThermoFisher). After fixation and permeabilization, cells were stained using Phalloidin AF568, ConcanavalinA AF488, Hoechst, SYTO14, WGA AF555 (all ThermoFisher). Finally, cells were imaged on a Yokogawa CV8000 high-content microscope.
In vivo target engagement
Since baseline PDGFRβ phosphorylation was almost not detectable by immunoblotting, 300 μg PDGF-BB (R&D Systems) was administered by intratracheal (IT) delivery. Each study included negative (no PDGF-BB) and positive controls (PDGF-BB, no compound). Intensities were quantified using Image Study Lite 5.2, and the % inhibition was calculated.
Nebulized delivery
A DSI inhalation tower (Buxco) was used for nose-only nebulization. Rats were acclimated for 10–15 minutes with air before exposure to JNJ-PDGFRi-1 for 50–55 minutes. A run without rats was performed, and deposition on an in-line gravimetric filter was used to calculate delivered drug per minute to determine the required exposure time.
Human dose projection
A multi-compartmental PK model based on Hendrickx et al. 14 was used. Percent inhibition and lung concentrations from rat PK and target engagement (TE) studies were fit using a 3-parameter Emax non-linear regression model in Graphpad Prism (version 10.1.2). The E0 parameter was kept constant at 0 and the Emax and EC50 values were estimated. Human dose projections aimed for the same trough inhibition as the 400 mg oral imatinib dose from the IMPRES trial 8 and assumed 30% drug delivery to the deep lung.
Statistical analysis
Percent inhibition data were analyzed in GraphPad Prism version 10.1.2 using a one-way analysis of variance test followed by a post-hoc Dunnett’s test to identify treatments with significant differences versus the PDGF-BB only group (positive control). p-Values were indicated on the graph as *** p < 0.001.
Results
An ideal inhaled PDGFRβ inhibitor would have improved potency compared to imatinib to accommodate nebulized dosing limits, long duration of action in the lung to enable once or twice daily dosing, and limited systemic exposure. We describe the steps that were taken to achieve these properties below.
PDGFRβ in vivo half-life determination
Extended PDGFRβ inhibition can be achieved by maintaining a high drug concentration in the lungs or by generating a slow-dissociating drug. 15 The latter approach requires a long PDGFRβ half-life in vivo. As shown in Figure 1A, incorporation of deuterium into PDGFRβ isolated from rat lungs was saturable, increased with time, and was fitted to a model that estimated the in vivo half-life of PDGFRβ protein to ∼7 hours. While the half-life of PDGFRβ in human lungs is not known, the relatively fast turnover in rats led us to focus on identifying compounds with a relatively flat PK profile to provide continuous lung exposure.
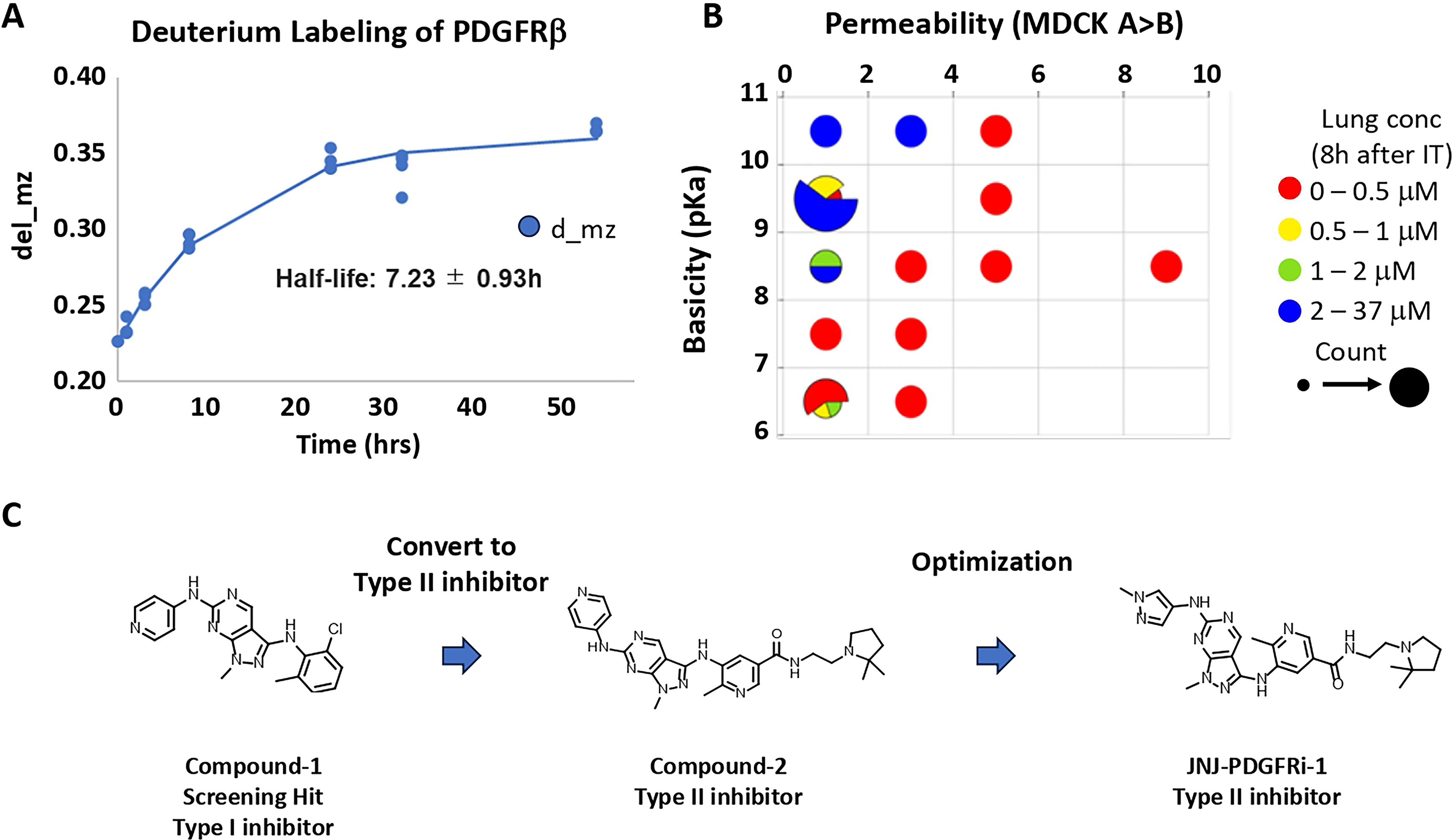
Physicochemical properties driving lung retention
Low permeability slows systemic absorption of an inhaled compound; however, sufficient permeability is required to enter target cells. Basicity was another important factor identified for lung retention, as basic compounds are often known to be lysosomotropic and associated with high pulmonary selectivity. 15 From the results of our 10K kinase-focused library screening for novel PDGFRβ inhibitors, through data mining and in silico virtual screening, we identified an initial 37 compounds that inhibit PDGFRβ and tested them in vivo. With these data points, we determined the critical values for maintaining lung retention: high basicity (pKa > 8) and permeability <4 (Fig. 1B).
Hit identification and optimization
Compound-1 was identified as an initial hit in the screen and based on its structure is predicted to bind to the ATP binding site (type I kinase inhibitor, Fig. 1C). To gain specificity, structure-based drug design was used to extend the molecule to make a hydrogen bond to the gatekeeper residue Thr681 and to interact with the allosteric back pocket (a type II kinase inhibitor). Imatinib similarly binds to both the ATP site and the allosteric back pocket. At the same time, a basic group was incorporated to improve lung retention (Compound-2). Several minor modifications of Compound-2 were then tested in vitro, leading to JNJ-PDGFRi-1. The pKa for JNJ-PDGFRi-1 is 9.2, while permeability is 0.85 × 10−6 cm/s.
JNJ-PDGFRi-1 is a more potent PDGFRβ inhibitor than imatinib
In vitro PDGFRβ kinase biochemical assays indicated that JNJ-PDGFRi-1 was a potent and selective PDGFRβ inhibitor; with pIC50 = 9.46 ± 0.04 in the PDGFRβ HTRF assay (Supplementary Fig. S1A) and pIC50 = 8.60 ± 0.05 in the PDGFRβ Lantha BND assay (Supplementary Fig. S1B). A pIC50 of 7.91 ± 0.06 was measured in the cellular A10 HTRF assay (Fig. 2A). In each assay, JNJ-PDGFRi-1 was more potent than imatinib. Finally, JNJ-PDGFRi-1 was inactive in the vascular endothelial growth factor receptor 2 (VEGFR2) ADP-Glo assay at the same concentrations used for the PDGFRβ biochemical and cellular assays (Fig. 2A).
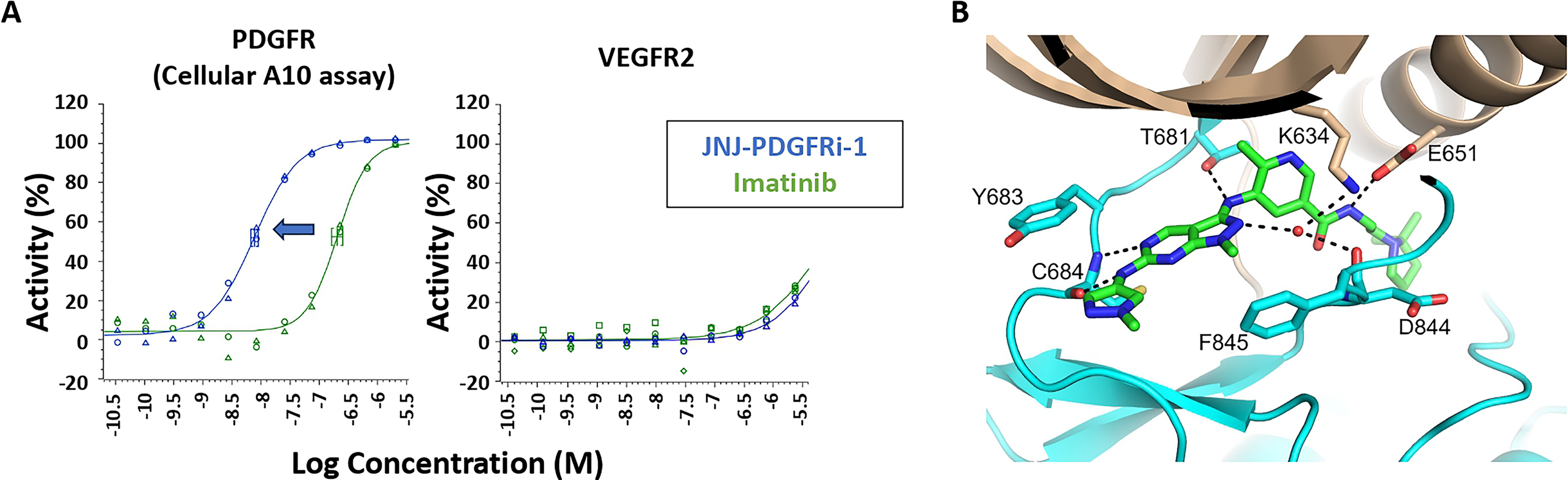
PDGFRβ crystal structure
To the best of our knowledge, no crystal structure has been described that fully covers the PDGFRβ kinase domain. Conditions were optimized to generate a co-crystal structure of PDGFRβ bound to JNJ-PDGFRi-1. As shown in Figure 2B, JNJ-PDGFRi-1 makes extensive contacts with PDGFRβ in the orthosteric ATP binding pocket. Specifically, the methyl-pyrazolopyrimidine of JNJ-PDGFRi-1 hydrogen bonds with the backbone nitrogen of Cys684. In addition, the small molecule makes water-mediated interactions with the backbone carbonyl oxygen of Asp844 and the side chain of Lys634. The nitrogen between the methyl-pyrazolopyrimidine and the methyl-pyrazole hydrogen bonds to the backbone carbonyl oxygen of Cys684, and the amide nitrogen between the methyl-pyrazolopyrimidine and the methylpyridine interacts with the side chain of Thr681. The amide nitrogen between the methylpyridine and the dimethylpyrrolidine interacts with the side chain of Glu651, while the oxygen interacts with the backbone nitrogen of Asp844 in the DFG-loop. The dimethylpyrrolidine sits in the back pocket and completely displaces the juxtamembrane domain.
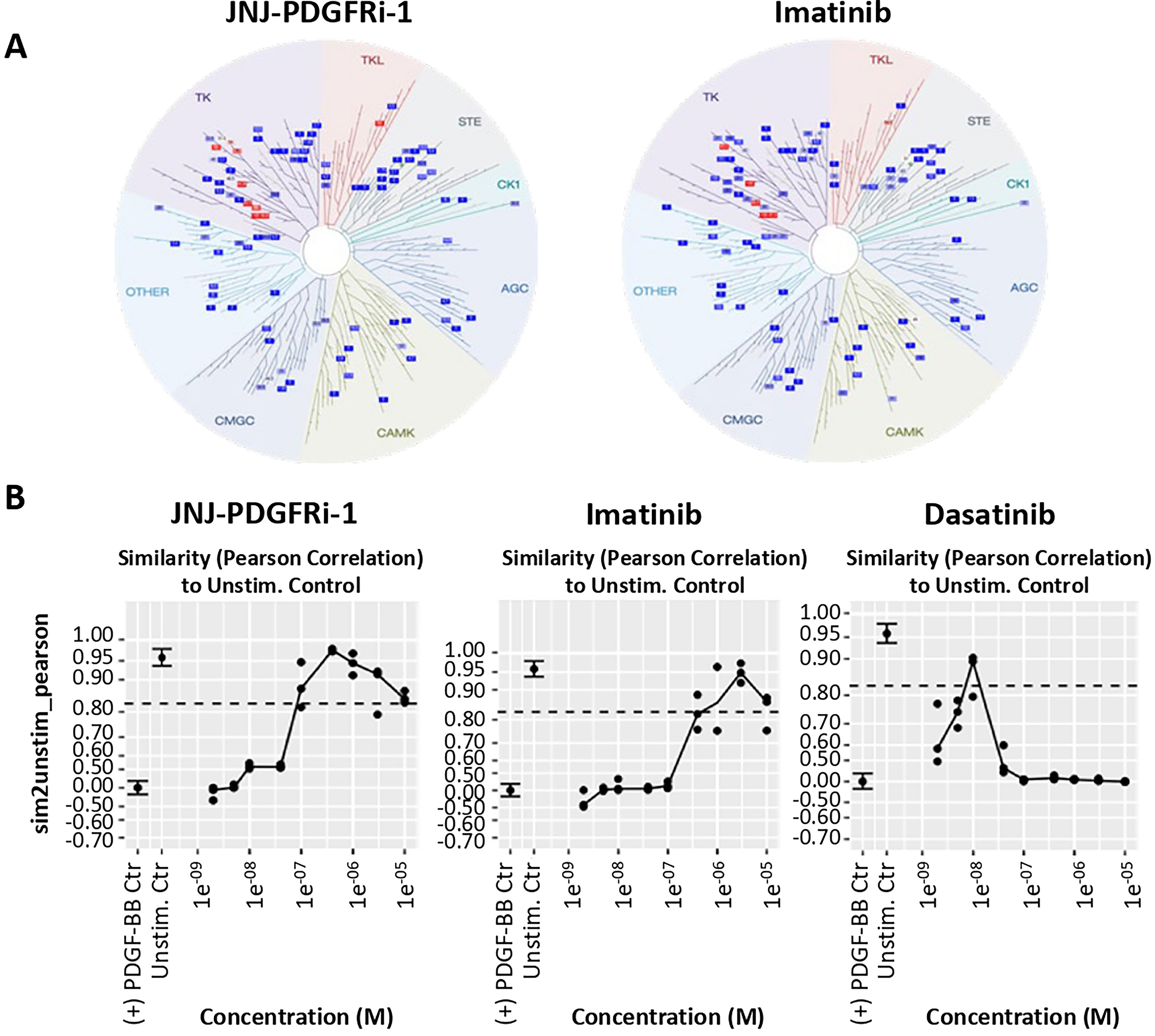
Kinase selectivity
Kinase selectivity was screened in a DiscoverX kinase panel that covers 70% of the human kinome. Overall, the RTK inhibitor profile of JNJ-PDGFRi-1 was similar to imatinib (Fig. 3A). For hits that showed >80% inhibition at 10 μM, binding affinity was confirmed in a second assay (Table 1).
Binding Affinity of JNJ-PDGFRi-1 to Selected Kinases

Kd, dissociation constant.
Cell painting high-content imaging
Cell painting is a target-agnostic, high-content imaging assay that uses multiplexed fluorescent dyes to assess changes in cellular morphology. 13 We tested whether compounds could block morphological changes associated with PDGF-BB treatment without inducing any non-specific changes that could indicate unwanted effects. As controls, imatinib and dasatinib were included. Like imatinib, dasatinib is an RTK inhibitor designed to inhibit the BCR-ABL fusion gene found in chronic myeloid leukemia (CML) patients but with a broader target profile 16 and increased risk to develop PAH. 17 The phenotypic similarity was quantified as the Pearson correlation between cells stimulated with PDGF-BB plus inhibitor and the unstimulated control (dimethyl sulfoxide-treated), which allowed for readouts of potency (lowest effective concentration [LEC]) and effective range (phenotype within 3-sigma of the untreated control). The JNJ-PDGFRi-1 LEC was 0.080 μM with an effective log range of 2.1, while imatinib had a LEC of 0.48 μM (Fig. 3B). The LEC of dasatinib was 0.0074 μM, but at greater concentrations, the phenotype differed from the unstimulated control. Importantly, JNJ-PDGFRi-1 and imatinib produced similar phenotypes over a broad concentration range, while differentiating from dasatinib (Fig. 3B).
DMPK properties and lung selectivity
The PK of JNJ-PDGFRi-1 in rats and dogs was characterized by a high plasma clearance (61 and 23 mL/min/kg, respectively) and a high volume of distribution at steady state (18 and 12 L/kg, respectively). The terminal half-life (T1/2) was 9 hours in rats and 14 hours in dogs; however, the mean residence time was shorter (3 hours in rats and 6 hours in dogs). After oral administration of JNJ-PDGFRi-1 (5 mg/kg in rats and 2 mg/kg in dogs), compound concentration was not measurable except at one early time point, indicating negligible oral bioavailability. The minimal oral bioavailability would limit the systemic exposure to JNJ-PDGFRi-1 from ingestion.
Following IT administration of JNJ-PDGFRi-1 (1 mg/kg) formulated as a solution, peak lung concentration was observed at 0.5 hours, the first sampling time point. The lung exposure was higher following IT administration, compared to IV administration, while the plasma exposure was comparable. The area under the curve ratio of lung over plasma was ∼5× higher following IT than following IV, indicating PK pulmonary selectivity (Fig. 4A).
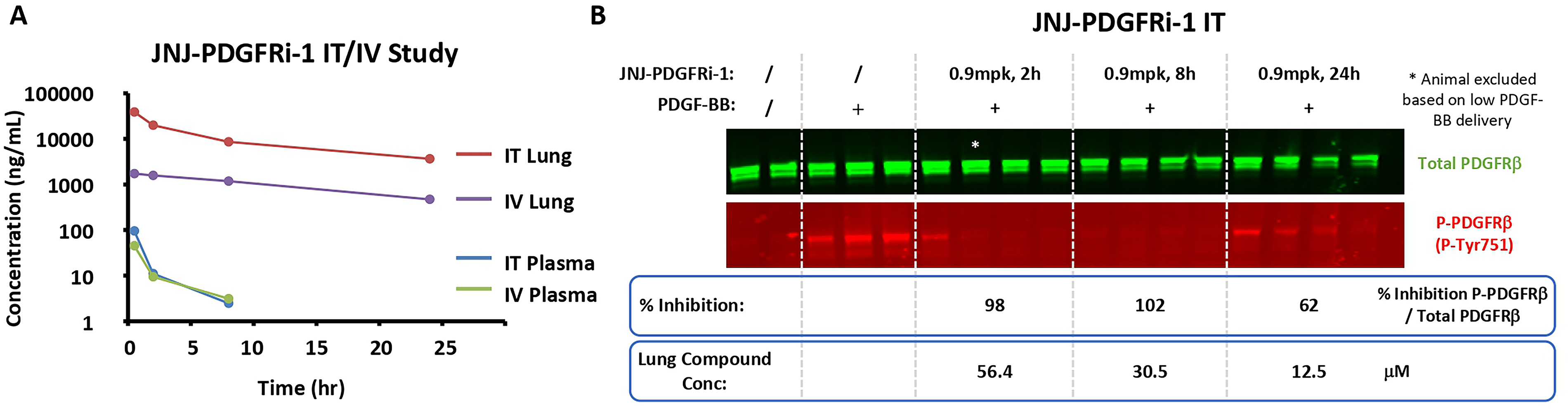
In vivo target engagement
A target engagement assay was developed where a dose of 300 μg PDGF-BB was delivered 5 minutes prior to takedown to activate PDGFRβ phosphorylation. IT delivery of JNJ-PDGFRi-1 at a 0.9 mg/kg dose showed almost complete inhibition of PDGFRβ phosphorylation for up to 8 hours. Even after 24 hours, 62% inhibition of phosphorylation was observed (Fig. 4B).
Formulation development for nebulized delivery
Developing a nebulized formulation requires careful assessment of solubility, formulation pH, viscosity, and stability.18,19 The pH-dependent solubility profile of the crystalline, dihydrate, free form is highest at low pH and decreases dramatically from pH 7 to 7.5 (Fig. 5A). Formulation development was further complicated by changes in viscosity across the preferred pH range of 5–7.25. A sharp increase in viscosity was measured between a pH of 5 and 6 and the formation of a semi-solid gel was visually observed.
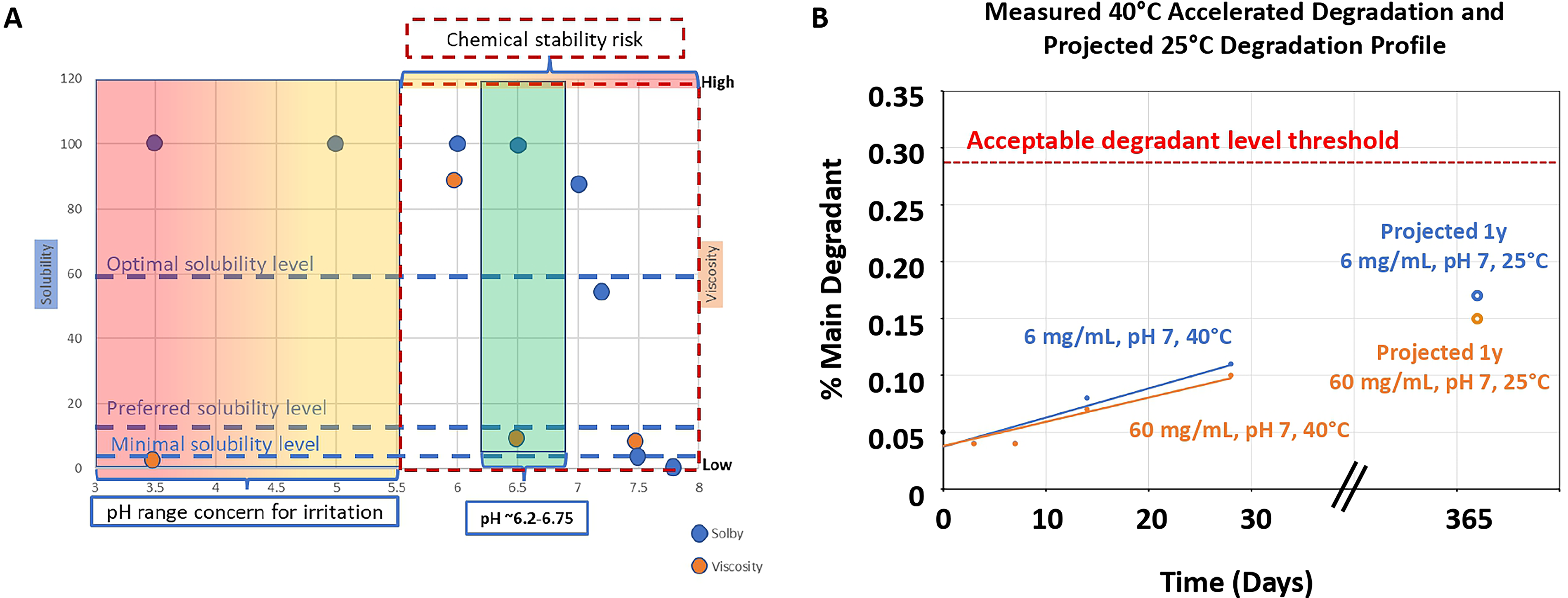
Stability was assessed using different buffers at 60°C. These studies showed an increase in chemical instability of JNJ-PDGFRi-1 between a pH of 6 and 8 (see Table 2). Accelerated stability studies were conducted at a maximum pH of 7 and at 40°C. Using Arrhenius kinetics, projections of shelf life can be established. 20 The data suggest that a formulation with a pH < 7.0 would likely achieve an initial ambient shelf life of at least 1 year (Fig. 5B).
Twenty-two hours Accelerated Buffer Stability of JNJ-PDGFRi-1 at 60°C in Various pH’s

Based on the compilation of data from solubility, viscosity, and stability studies, an optimal pH window for a nebulized formulation of JNJ-PDGFRi-1 was identified to be between 6.2 and 6.75.
Nebulized delivery of JNJ-PDGFRi-1
A cascade impactor was used to measure deposition of particles as a function of their size (Fig. 6A). The corresponding mass median aerodynamic diameter values were well below 5 μm, indicating a small size that allows for particle deposition into the deep lung. 21 Next JNJ-PDGFRi-1 was delivered by nebulization to rats.
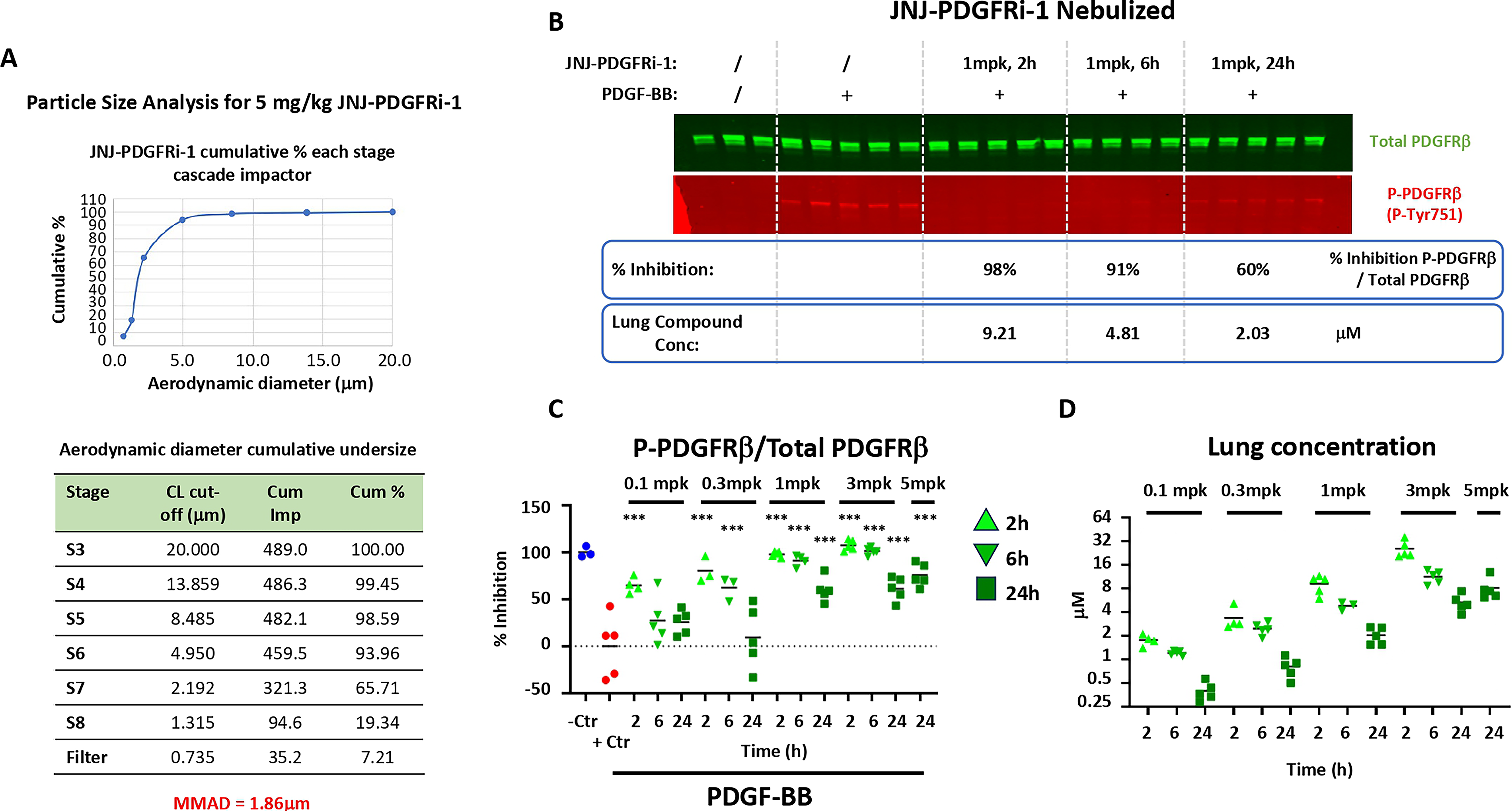
Phosphorylation of PDGFRβ was inhibited in a dose- and time-dependent manner by JNJ-PDGFRi-1. For example, 98% inhibition was seen 2 hours after a 1 mg/kg dose and 60% after 24 hours (Fig. 6B). Results from a time and dose response are summarized in Figure 6C. In parallel, plasma and lung PK of JNJ-PDGFRi-1 were measured. Lung exposure increased in a roughly dose-proportional manner (Fig. 6D, Table 3).
Single Dose Pharmacokinetics of JNJ-PDGFRi-1 in Rats Following IT Administration or Nebulized Inhalation

Values are mean or mean ± standard error. n = 3 animals/time point.
AUC, area under the curve; IT, intratracheal; IV, intravenous; NC, not calculated.
Human dose projection and PK/PD relationship
A schematic of the multi-compartmental lung PK model is provided in Figure 7A. The rat PK parameters were estimated by fitting the IV/IT and nebulized PK data sets. From the nebulized TE study (Fig. 6), lung concentrations were plotted against % inhibition (Fig. 7B), and the in vivo IC50 was calculated using a static approach, which resulted in estimates of EC50 (1200 ng/mL) and Emax (120%).
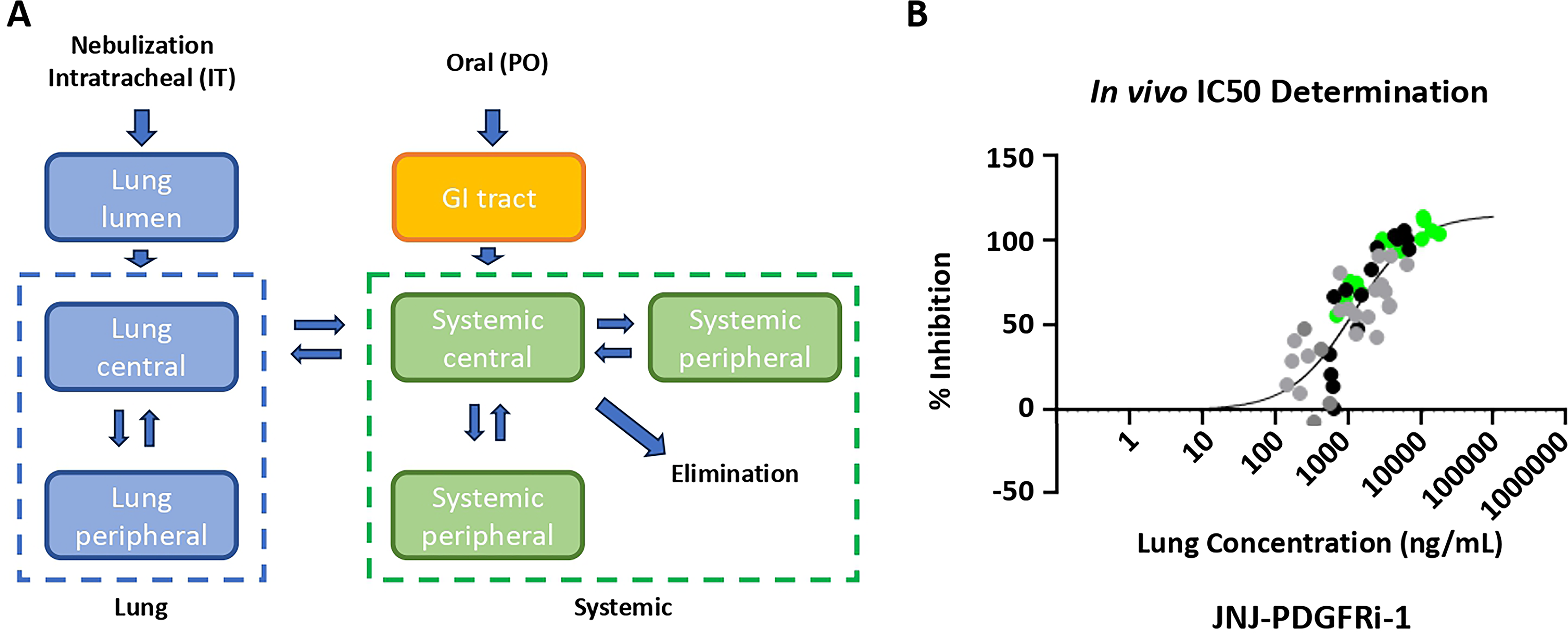
The PK and TE parameters were scaled to human PK parameters, and the human steady-state PK was simulated for both QD and BID administration. In order to match oral imatinib inhibition at trough at steady state 22 40 mg daily or 12 mg twice daily of JNJ-PDGFRi-1 is needed. The Ctrough approach was used as it was the most conservative approach.
Discussion
JNJ-PDGFRi-1 showed extended lung retention and improved potency compared to imatinib, which minimizes the required dose to maintain continuous inhibition of PDGFRβ. The kinase inhibition profile of JNJ-PDGFRi-1 matches imatinib, and minimal activity against VEGFR2 was accomplished by a careful structure-based design. Conditions were identified that resulted in a small nebulized particle size, which allowed for deep lung deposition. The projected human dose was 12 mg twice daily. A schematic summary of optimized attributes is provided in Figure 8.
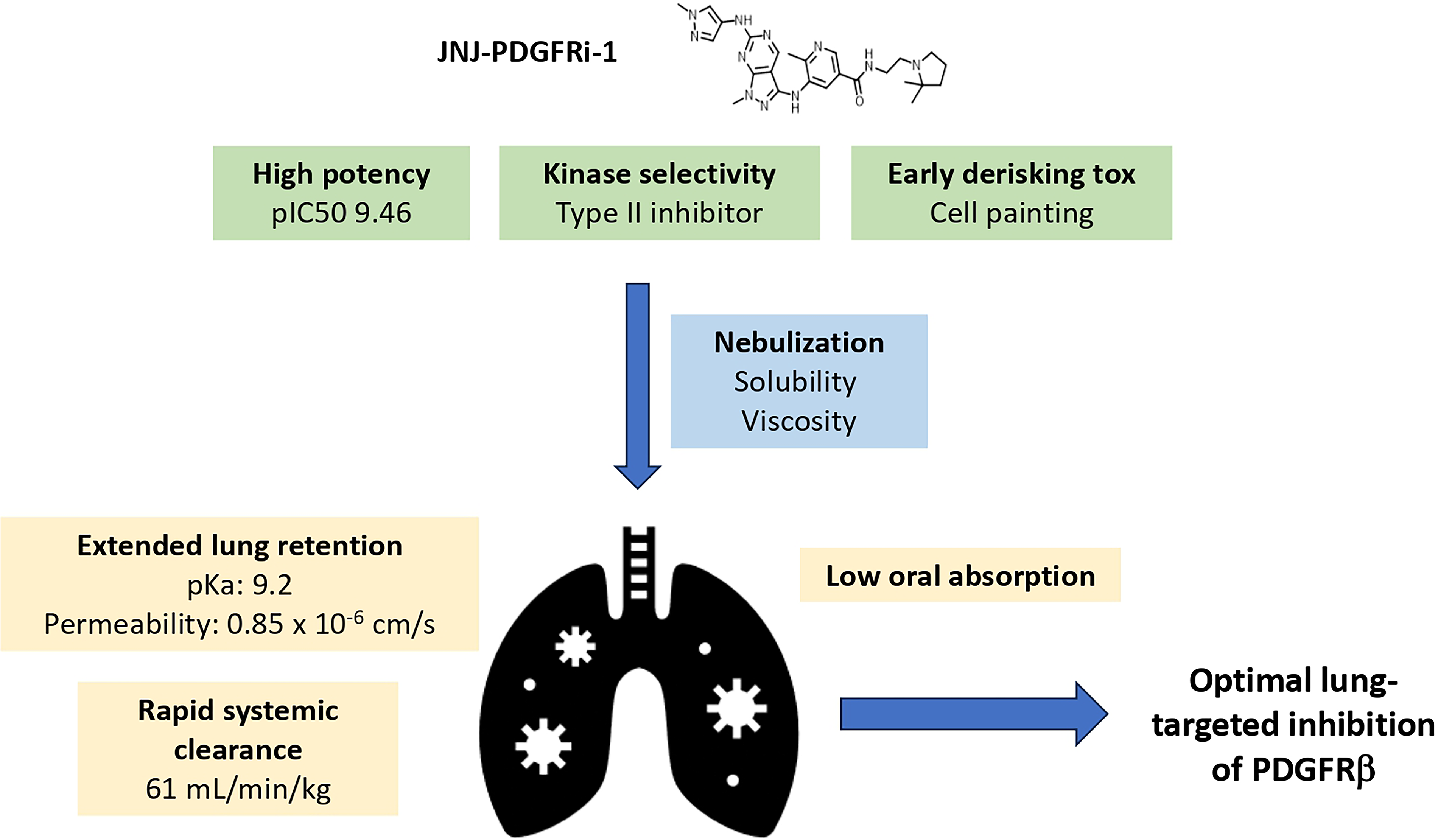
Schematic representation of JNJ-PDGFRi-1 attributes that were optimized to achieve optimal lung-targeted inhibition of PDGFRβ.
Safety considerations were paramount in our screening strategy. The cell painting assay provides an unbiased way to look at possible alterations in cellular morphology and function. We set this assay up in a smooth muscle cell line, which expresses our target RTK, but for other RTKs other cell lines could be used. At the same time, we used selectivity over VEGFR2 as a counter screen because deletion of VEGFR2 signaling in endothelial cells resulted in mild pulmonary hypertension in mice, 23 heterozygous loss of function of VEGFR2 is associated with the development of PAH, 24 and VEGFR2 inhibition combined with chronic hypoxia results in complex vascular lesions in rats. 25 Furthermore, bevacizumab, a VEGFR2 inhibitor, increases the risk of developing PAH. 26
Depending on the specific RTK, some modifications to our screening and optimization strategy are likely needed. For example, for receptors with a long in vivo half-life, the screening strategy could focus on identifying compounds with a slow off-rate. While JNJ-PDGFRi-1 has a high affinity for PDGFRβ, it also inhibits other highly related RTKs (Fig. 3A, Table 1), similar to imatinib. 27 While the ATP-binding site is highly conserved across different RTKs, differences in the surrounding protein can be used to enhance compound specificity (type II kinase inhibitor). For other RTKs, the closest related RTK with a potential safety risk should be identified. For PDGFR, almost no phosphorylation was detected at baseline, which required us to deliver the PDGF-BB ligand to induce phosphorylation in order to measure inhibition of PDGFR in vivo. For other RTKs, this may not be needed, and it is recommended to implement higher throughput target engagement assays if they are available for a specific RTK. Finally, optimization of nebulization conditions will be highly compound-specific, and our recommendation is to perform pH-dependent solubility, viscosity, and stability measurements as early as possible during the discovery program to eliminate compounds that would be difficult to nebulize.
For in vivo testing of JNJ-PDGFRi-1, our tested doses and delivery methods changed depending on the stage of the program. The rationale for this was that a relatively low dose of 0.9 mg/kg dose delivered by IT instillation allowed us to differentiate compounds (Fig. 4B). As we started focusing on JNJ-PDGFRi-1, we transitioned to nebulized delivery. Drug delivery efficiency is lower for nebulization, and given this uncertainty, we tested a wider concentration range (Fig. 6B).
Recently, no significant benefit was seen in PAH patients treated with an inhaled dry powder formulation of imatinib (phase 2b IMPAHCT trial 28 ). This result underscores the need for additional work to identify compounds that are tailored for lung retention and continuous inhibition of RTKs in the deep lung.
In conclusion, this article provides a roadmap for the development and de-risking of highly potent inhaled compounds with extended activity in the lungs. This approach could be used for different RTKs across several pulmonary diseases, including PAH, COPD, asthma, and idiopathic pulmonary fibrosis.
Authors’ Contributions
G.M., Z.H., M.E., C.H.H., A.D.R., S.I., V.W., M.H., S.J., Y.S., P.S., P.A., D.D., J.H.C., E.D., W.W., D.R., and L.M.Y.: Acquired data. G.M., T.L., Z.H., K.H., M.E., C.H.H., A.D.R., S.I., V.W., M.H., S.J., Z.L., Y.S., P.S., P.A., D.D., J.H.C., E.D., W.W., H.B.T., D.R., S.P., S.P., S.A.H., A.R.N., L.M.Y., A.S.S., C.J., D.K., S.X., and D.R.B.: Analyzed and interpreted data. G.M., T.L., and D.R.B.: Drafted article. Z.H., K.H., M.E., C.H.H., A.D.R., S.I., V.W., M.H., S.J., Z.L., Y.S., P.S., P.A., S.A.H., A.R.N., L.M.Y., A.S.S., C.J., D.K., and S.X.: Contributed to article.
Footnotes
Acknowledgments
The authors would like to acknowledge the contributions of Tianbao Lu, who passed away on March 14, 2025. He was a driving force behind this article and contributed his medicinal chemistry insights to numerous small molecule therapeutics. He was a friend, loving husband, and father and will be sorely missed by his colleagues at
The authors would also like to acknowledge Neetu Shukla and Marcy Gvazdauskas for help with analyzing drug deposition in the cascade impactor and the biotechnicians that supported the animal studies.
Disclosure Statement
T.L., Z.L., and D.R.B. are inventors on a patent application filed by Actelion Pharmaceuticals that is related to the molecule described in this article. Other authors declare that they have no competing financial or nonfinancial interests.
Funding Information
All studies were funded by
Supplemental Material
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.